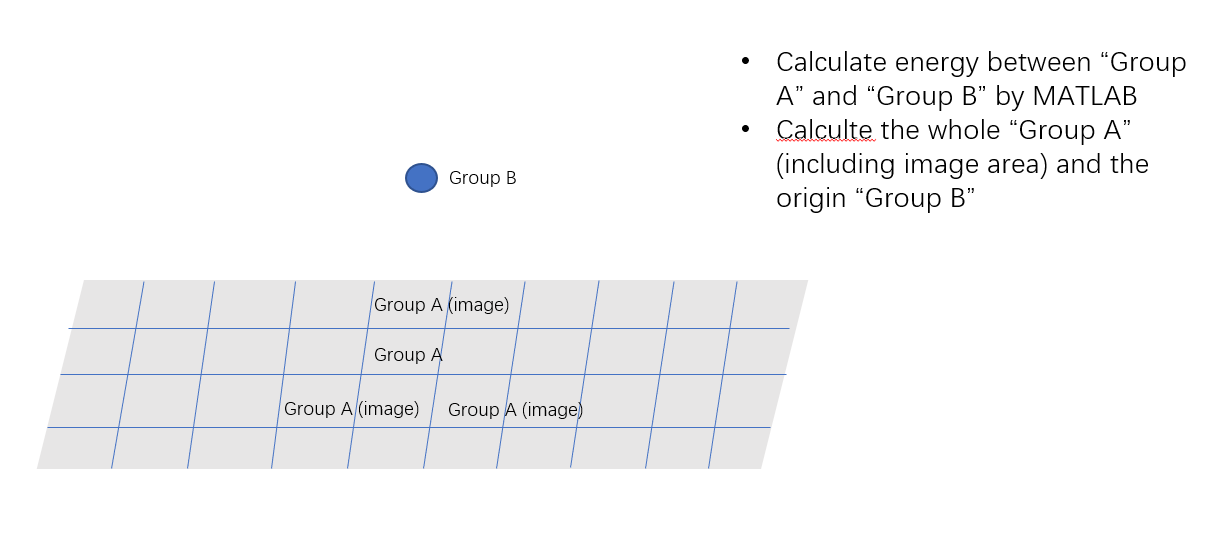
I have used “compute group/group” command to calculated energy between “Group A” and “Group B”(including “Ksapce_style ewald” in 3D-periodic box).
There is a question that what the final output from Lammps means? Does it means the energy between the “Group A” and “Group B” in the infinite region(including all image area) or only the origin box area(calculate the whole image area, and than average it and output)? Both “Group A” and “Group B” have the net charge.
And, I also calculate the energy by the Matlab, but I find its result is much larger than lammps’s, I don’t know why. The calculated scheme is attached.
I have used “compute group/group” command to calculated energy between “Group A” and “Group B”(including “Ksapce_style ewald” in 3D-periodic box).
There is a question that what the final output from Lammps means? Does it means the energy between the “Group A” and “Group B” in the infinite region(including all image area) or only the origin box area(calculate the whole image area, and than average it and output)? Both “Group A” and “Group B” have the net charge.
it is the same computation as when you normally compute forces and energies, so that you should get the same energy for pairwise and long-range coulomb energy when both groups are “all” (minus bonded interactions). that includes interactions with any periodic copies or the closest periodic image for the real-space pairwise interactions and the interactions with all periodic copies of the two interacting groups. for the kspace contribution there is even a PDF document linked from the manual page of compute group/group that describes how the kspace contribution is computed.
And, I also calculate the energy by the Matlab, but I find its result is much larger than lammps’s, I don’t know why. The calculated scheme is attached.
most likely your matlab calculation didn’t properly account for periodic boundary conditions and cutoffs.