Hello.
- With regard to your first question, I posted a similar question some time back.
This would require unwrapped coordinates to be available as variable.
Dear Steve: Is it true that the unwrapped variables are available only for dump, but not as a variable.
Is there any way to access unwrapped coordinates as a variable?
Another problem is that the PE/atom does not include electrostatic contributions. So this problem still remains.
-
Regarding your second point: I too would appreciate if thefix_heat_flux.cpp be modified to include the modifications made by German. The pe/atom could also be modified based on Germans additions. This decision however has to be taken by Steve and the others managing the code.
-
Dear German,
Attached with this mail is an input file in.lj.compute, that combines both the input files you had posted ie using variables and using compute heat/flux. For testing, I have significantly reduced the timesteps.
Here are a few values from the flux.log file:
Step Temp Press TotEng PotEng flux[1] flux[2] flux[3] Jx Jy Jz Volume
0 40.797567 -100.90087 -63.724713 -68.275743 -1.2503741 0.35527064 -2.0326868 -1.2503741 0.35527064 -2.0326868 33823.124
1 40.860126 -102.07204 -63.724594 -68.282602 -1.2789893 0.32576403 -2.0379642 -1.267529 0.35131522 -2.0140116 33823.124
2 40.928747 -103.40437 -63.724594 -68.290258 -1.2958122 0.32209609 -2.0182231 -1.284414 0.34793316 -1.9940625 33823.124
3 41.003351 -104.87917 -63.724528 -68.298514 -1.3115363 0.318944 -1.9981188 -1.300198 0.34505271 -1.9737588 33823.124
4 41.083841 -106.50637 -63.724516 -68.30748 -1.3272088 0.31663561 -1.9776122 -1.3159291 0.34300364 -1.9530632 33823.124
5 41.170105 -108.25633 -63.724264 -68.316851 -1.3420114 0.31536598 -1.9562788 -1.3307894 0.34198112 -1.9315504 33823.124
As you can see, apart from the first step, the flux values differ slightly.
I agree that the thermal conductivity values using the two different sets agree closely.
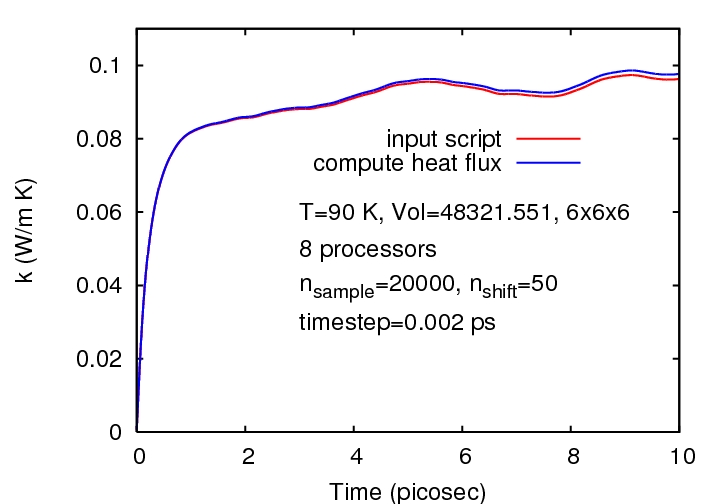
However, I feel it would be nice to know what causes the difference in values, since both are trying to use the same formula and calculate exactly the same quantity. Also, the deviation in thermal conductivity values calculated by the two methods increases as the time increases (ie as the nuber of points over which averaging is done decreases) (in fig LJ_90K.jpg).
Do you have any clues as to what is causing this difference in values?
I haven’t been able to set up the run to calculate the thermal conductivity of water, including long range effects with the codes you have sent. I shall try to complete that soon and shall post the results.
Thanks and warm regards,
Mario
in.lj.compute (1.78 KB)
log.lammps (18.2 KB)
flux.log (4.86 KB)