As the title suggests, I would like to compute the pressure of a certain group of granules in a specific region of my domain. My simulation involves compressing granules (type 1) with a wall made out of smaller granules (type 2) on the boundary. I would like to perform this computation over the compressed granules (type 1) during the evolution of the compression. For now you can assume my domain of interest where the granules are present to be a rectangle. I have been currently doing this using the commands: region, group region and then compute stress/atom with compute reduce sum. However, I think this won’t stop the small granules (type 2) to being a part of my calculation. Is there a way I can confirm this or make changes to the way I am computing this?
My snippet is:
region atom_region block 0.1 0.3 0.1 3.9 -1e7 1e7
group atoms region atom_region
compute peratom atoms stress/atom NULL pair
compute p atoms reduce sum c_peratom[1] c_peratom[2] c_peratom[3]
I think the problem lies in the first compute command as it takes all granules irrespective of the type as long as it in the specified region.
there are multiple ways to compute the sum over the intersection of the region and atom type:
define a group by type and then use compute reduce/region
define a group by type and a second group by region and then apply compute stress/atom on the first and compute reduce on the second
define a group by type and a second group by region and a third group as the intersection of the two and then apply compute stress/atom and compute reduce to that group
that still leaves you with the challenge of determining the area of the intersection to convert from stress to pressure.
I forgot to cc everyone in the reply (my apologies Axel!)
Thanks for the prompt reply. I was wondering if this will also solve the problem relating to the second email I sent (my apologies for not including this in the main email), that is will it exclude the pair-waise interaction between type 1 and type 2. This is because I want to find the ‘local pressure’, and I would like it to be based on the stress due to the internal forces that are wholly due to type 1. This problem was addressed in this thread in the archive but I don’t see a solution:
I forgot to cc everyone in the reply (my apologies Axel!)
Thanks for the prompt reply. I was wondering if this will also solve the problem relating to the second email I sent (my apologies for not including this in the main email),
there was no second e-mail today, so what e-mail is this in reference to?
are you the original poster (despite using a different name and e-mail address)?
that is will it exclude the pair-waise interaction between type 1 and type 2. This is because I want to find the ‘local pressure’, and I would like it to be based on the stress due to the internal forces that are wholly due to type 1. This problem was addressed in this thread in the archive but I don’t see a solution:
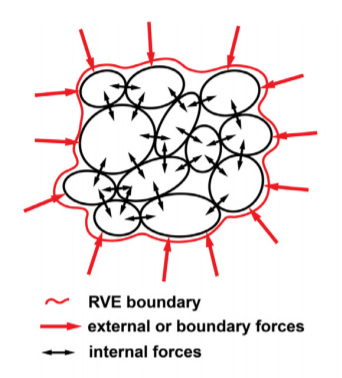
Sincere apologies. I replied using my other email address. I was referring to the attached figure. Is there any way I can calculate the virial stress for the subvolume of the black granules only. That is accounting for the black arrow forces throughout the entire volume excluding the red externally applied forces? Say the black granules are the main granules of interest to me and the boundary is the applied forcing.
Thank you very much and apologies for the confusion.
Sincere apologies. I replied using my other email address.
please use only one e-mail address and only one name. showing up with “multiple personalities” is quite confusing and thus counter-productive.
I was referring to the attached figure. Is there any way I can calculate the virial stress for the subvolume of the black granules only. That is accounting for the black arrow forces throughout the entire volume excluding the red externally applied forces? Say the black granules are the main granules of interest to me and the boundary is the applied forcing.
I think I had pointed out where to find the answer already, but - if need be - here is more detail. the per-atom stress information is collected during the force computation and thus you cannot change what contributes outside of the categories available on the compute stress/atom command. in other words, you can include or not contributions from pair style interactions or bonded interactions for fixes, but you cannot partition the per-category contribution without changing those forces.