Dear LAMMPS users,
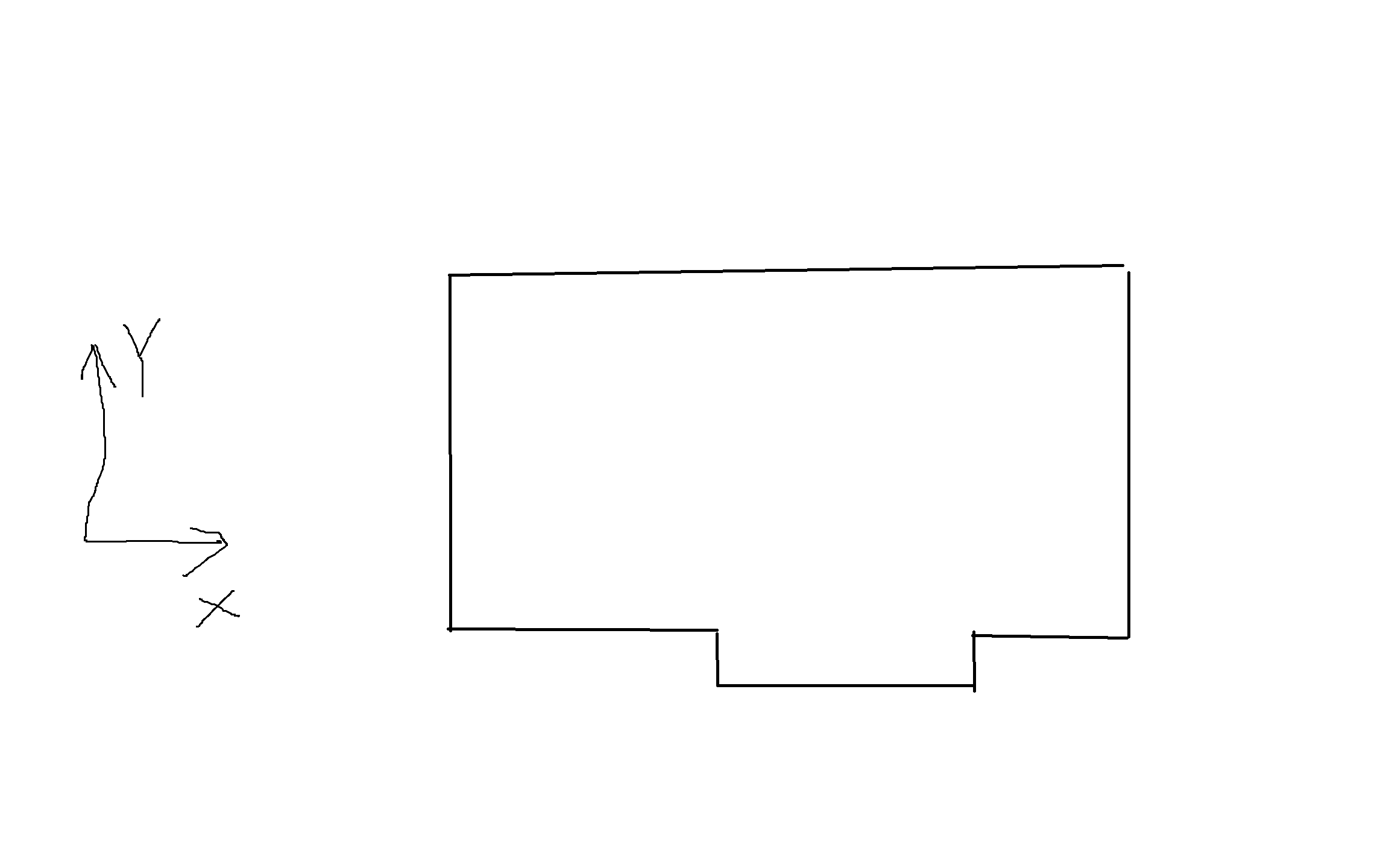
I want to create a groove in the simulation box as the picture presented. The system is 2D. The lines of code I have used are:
region reg block 0 100 0 50 -0.5 0.5 units box
region hole block 30 80 -10 -1 -0.5 0.5 units box
region combined union 2 reg hole units box
create_box 1 combined
In LAMMPS, simulation boxes must be parallelepipeds (also called “prisms”). The figure you drew is not a prism. From the documentation:
“If the region is not of style prism, then LAMMPS encloses the region (block, sphere, etc) with an axis-aligned orthogonal bounding box which becomes the simulation domain.”
So LAMMPS is going to create a rectangular simulation box if you provide it with the region that you drew in the picture you included.
Instead, what it sounds like you are trying to do is confine your simulation inside a container with a non-rectangular shape. There are several ways to do this. You can try using the “fix wall” commands, but these work best if your container is convex. Since your container is not convex, I suggest arranging atoms uniformly on the surface of the container and then immobilizing them. You can do this using either the “read_data” or “create_atoms” command. If you use the “read_data” command, you must first create a LAMMPS “DATA” file. The basic DATA file format is explained here. Several programs to help you create DATA files are listed here. You can also create them manually, since the format is simple.
Then create a “group” of atoms which does not include the atoms on the surface of the container. Use that group with the fix that integrates the equations of motion (such as “fix nvt”, “fix nve”, etc…), instead of using the “all” group. This will immobilize those atoms.
The following examples use DATA files, along with the “read_data” command (instead of “create_atoms”). The DATA files define the geometry of the system, bonded topology, as well as the positions of the atoms in the container wall. (The “.data” files in those examples must be created by a program called “moltemplate.sh”. However you can ignore the moltemplate-specific details. Just read the the “run.in.nvt” and “run.in.min” files for more details. You can read and understand these files without being familiar with moltemplate.)
- General discussion here.
Particles confined in a pentagonal cylinder (picture here).
-
particles and polymers confined in a 3D ellipsoid (picture here)
-
Coarse grained polymer confined within a container of arbitrary shape.
The example is located here. Unfortunately this is a complicated example.
Andrew
P.S.
Details for example #3: The following details are only relevant to moltemplate users. The moltemplate files and other files for examples 1 and 3 are here and here, respectively. In the first example, a flat planar wall is created and copied and rotated 5 times to create a pentagonal cylinder. In the 3rd example, the container has an arbitrary shape. The positions of the atoms in the container wall are stored in the “capsid_locations.txt” file. In this example, that file is (STEP1) renamed to “coords_wall.raw” and then (STEP2) converted into moltemplate LT format (“wall.lt”), and finally converted into LAMMPS DATA format using the “moltemplate.sh” program.
Thanks for the help, I have seen the examples and it was helpful.
{kind=link}