Dear LAMMPS developers
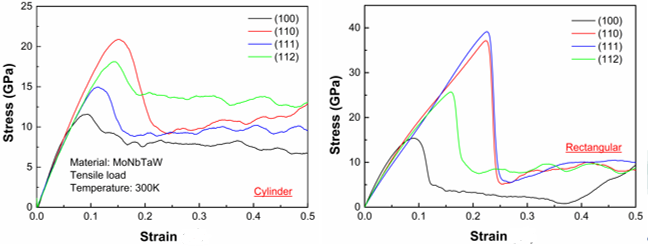
I am Jing Guo from UK. I have been working on the MD simulation for high entropy alloys recently. I carried out the simulation of tensile process for NbMoTaW HEA. I built cylinder and rectangular models with similar size of Φ60×240Å and 60×60×240Å considering different orientations, the code for both of these two models is exactly the same. However, the results for these two models are different from each other. Please see attached file. In addition, there is no necking and fracture happened in the simulation even the strain value is 0.5. The lammps code has been listed below. Could you please tell me is there something wrong with my code? What caused the results different from each other after I changed the shape of model?
Input:
#Initialization#
units metal
atom_style atomic
boundary p p p
neighbor 0.3 bin
#create geometry#
read_data NbMoTaW110.lmp
#define interatomic potentail#
pair_style snap
pair_coeff * * MoNbTaW.snapcoeff MoNbTaW.snapparam Ta Nb Mo W
#Output thermodynamics information#
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
compute 1 all stress/atom NULL
compute 2 all ke/atom
compute 3 all displace/atom
#Output#
dump 1 all custom 500 tensile.dump id type x y z
#Relax structure#
velocity all create 300 12345 mom yes rot no
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
#Run#
timestep 0.001
run 20000
#Tensile process (Z direction)#
unfix 1
fix 1 all npt temp 300 300 1 x 0 0 1 y 0 0 1 drag 1
fix 2 all deform 1 z erate 0.01 units box remap x
variable Ltmp equal lz
variable Lini equal ${Ltmp}
variable strain equal (lz-v_Lini)/v_Lini
variable stress equal -pzz/10000
fix 3 all print 100 “{strain} {stress}” file strain_stress.txt screen no
#Run#
run 50000
Best regards,
Jing