lammps users:
Hi, every lammps users!
I’m doing DPD simulation.
Nowadays, I have met some confused things on dump command.
I compiled lammps with all packages (lammps-7Jul09, ubuntu 8.04, mpich–8) .
My example is di-block polymers, A5B5, 101010, 300 molecules, 3000 atoms.
My in script is as follows:
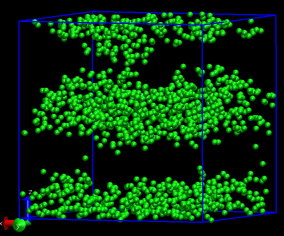

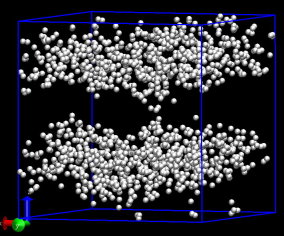
If you are asking why the dump files for dump "all" and dump "polymer"
are the same, then what was the output of this command:
group polymer type 1 2
Did it say the new group contained all atoms or only some of them?
Steve
Hi, Zhou Tao
My question is actually not concerned about your problem.
I would like to know these lines in your input script:
"
pair_coeff 1 1 25.0 4.5
pair_coeff 1 2 80.0 4.5
pair_coeff 2 2 25.0 4.5
"
does not in consist with the settings in Groot and Madden’s [J. Chem. Phys.,108,8713,(1998)] paper.
In their paper, the spring force is set as:
f=C__r__ij
while in your script it is:
f=C(r-ro)
this may considerably affect the results obtained~
I wonder is there any bond style in lammps is in the first form?
if not, how can I get this bond style work, write my own bond style?