Dear Lammps List,
I am simulating a system in which I keep part of the atoms rattling around their original positions with spring/self to make a nanopore.
The pore is filled with liquid.
I can’t get energy conservation for this system in NVE simulation even with a very short time step (0.1fs). The same system without
spring/self displays excellent energy conservation for this time step and much higher (up to 5-10 fs is ok).
The spring/self constant i use is quite low, 5 kcal/A2mol, so I am quite surprised the energy is not conserved and wonder if there may
be anything in the implementation of spring/self that prevents energy conservation.
I am attaching the input and auxiliary files. I would appreciate any help or advice on how to fix this.
thanks,
vale
in.nve-springwall (1.92 KB)
mW.sw (1.24 KB)
restart-nve-300.500000 (337 KB)
Hi Vale. What you’re seeing is not surprising. This fix does not conserve energy. The reason is because the fix imparts forces without tallying the corresponding contribution to the potential energy. I see no reason why code couldn’t be added that would keep track of this and hence allow observed energy conservation. The dynamics would not change.
Paul
Paul,
thanks for your answer. I do not know how difficult would be for the developers to add the contributions of the
springs to the total energy (or at least report it independently), if that’s not in lammps plans I’ll try to add it myself.
Nevertheless, there is something I still do not understand: If the problem is only reporting but not true energy
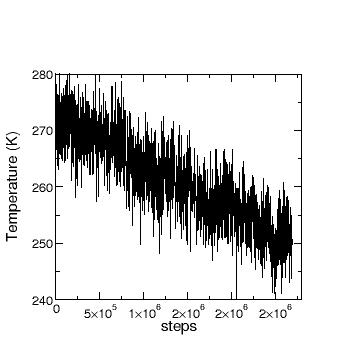
conservation, why does the temperature drop?
thanks for your help!
vale
Vale,
Although there is no immediate plan to add fix spring/self contributions to the total energy, I think that it would be a fairly minor task, so you might hold off for a week or so to see if we get to it before diving into it yourself.
The T drop you’re seeing is expected. Kinetic energy from the atoms gets stored as spring/self potential energy when the atoms move off of their initial positions. This spring/self potential energy is not currently being computed nor reported by LAMMPS. The loss of kinetic energy results in a temperature drop. If you really want to have constant T, you should run NVT rather than NVE, but then you won’t conserve energy.
Paul
Paul,
Thanks for your answer. I will wait to see if you [the developers] can add the spring energy to
the total. Nevertheless, I suspect the energy leaking is not related to this.
I am afraid there may be some additional origin for the lack of energy conservation because
is not that the energy oscillates due to an equilibrium exchange with the springs, but that it
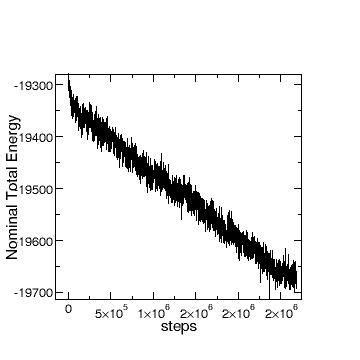
drifts. This may not have been clear in my previous email, so I include here the pictures of
“total” E vs step and T vs step.
I really appreciate your help on identifying the cause of this problem.
thanks!
vale
Vale,
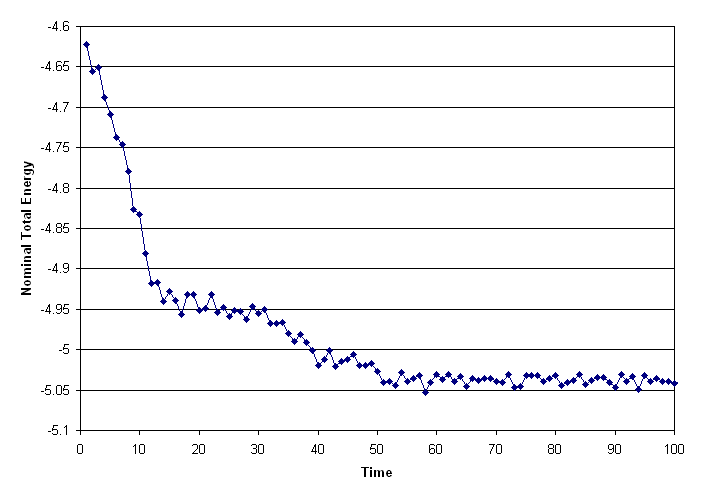
I suspect that if you run it out long enough, that the total energy will eventually plateau out. I ran a simple test of fix spring/self on a 256 atom LJ system, and found that there was total energy and temperature drop (as expected; see previous explanation), but if I ran it long enough, they plateau out. Use of a larger spring constant produces the plateau earlier. Here’s my simple test script:
3d Lennard-Jones melt
variable x index 4
variable y index 4
variable z index 4
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 $x 0 $y 0 $z
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 1.44 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify delay 0 every 20 check no
fix 1 all nve
fix 2 all spring/self 1.0
thermo 1000
run 100000
And I’ll attach the resultant energy plot. As expected, there is an initial loss of energy to the springs, then an equilibrium is reached.
Hopefully this helps.
Paul