Hi All,
I am trying to use the fix bond/create command to create bonds. My simulation starts with no bonds. Based on the LAMMPS manual, I need to use the bond style and bond_coeff to define bondtype. So I input the scripts below:
bond_style harmonic
bond_coeff * 110 1.0fix 2 all bond/create 10000 1 3 10 1
However, it shows an error below:
ERROR: Numeric index is out of bounds (…/bond_harmonic.cpp:128)
Can anyone help to give some suggestions on solving this problem?
Thanks
You have to reserve space for storing bonds and you have to declare how many bond types you want to have when creating the simulation box.
those values are locked in and cannot be changed after the simulation box is created.
Thanks for your help. Could you please give me some ideas on how to reserve space for storing bonds. Basically, my simulation has two data files with no bonds defined. If I want to reserve bonding space, should I input bond types in one of the data files?
please check out the documentation for the read_data command: https://docs.lammps.org/read_data.html
you will need to use the extra/bond/types, extra/bond/per/atom, extra/special/per/atom parameters with suitable values (extra/bond/types needs to be exact or you will have to provide (dummy) parameters for the unused bond types, the others can be larger than needed).
in general, i would recommend to read in both data files, create the bonds, and then write out a new data file with everything.
then you can use that combined data file without any “extra” keywords as LAMMPS will detect the optimal settings when reading it.
axel.
Hello,
I have added extra/bond/types in my first read data file. However, the same problem still happens
Basically, I have two data files with no bonds defined, so I set the data files like below:
read_data 1.data extra/atom/types 1 extra/bond/types 1 extra/bond/per/atom 4 extra/special/per/atom 4
read_data 2.data add append offset 2 0 0 0 0
After adding potentials for all the atoms, I set the bonds like below.
bond_style harmonic
bond_coeff 1 100 1.1
I set the bond type number as “1” because I only create one new bond. However, the system still shows that the numeric index is out of bound. COuld you please give me some ideas what the errors could be?
Thanks
please provide your screen output
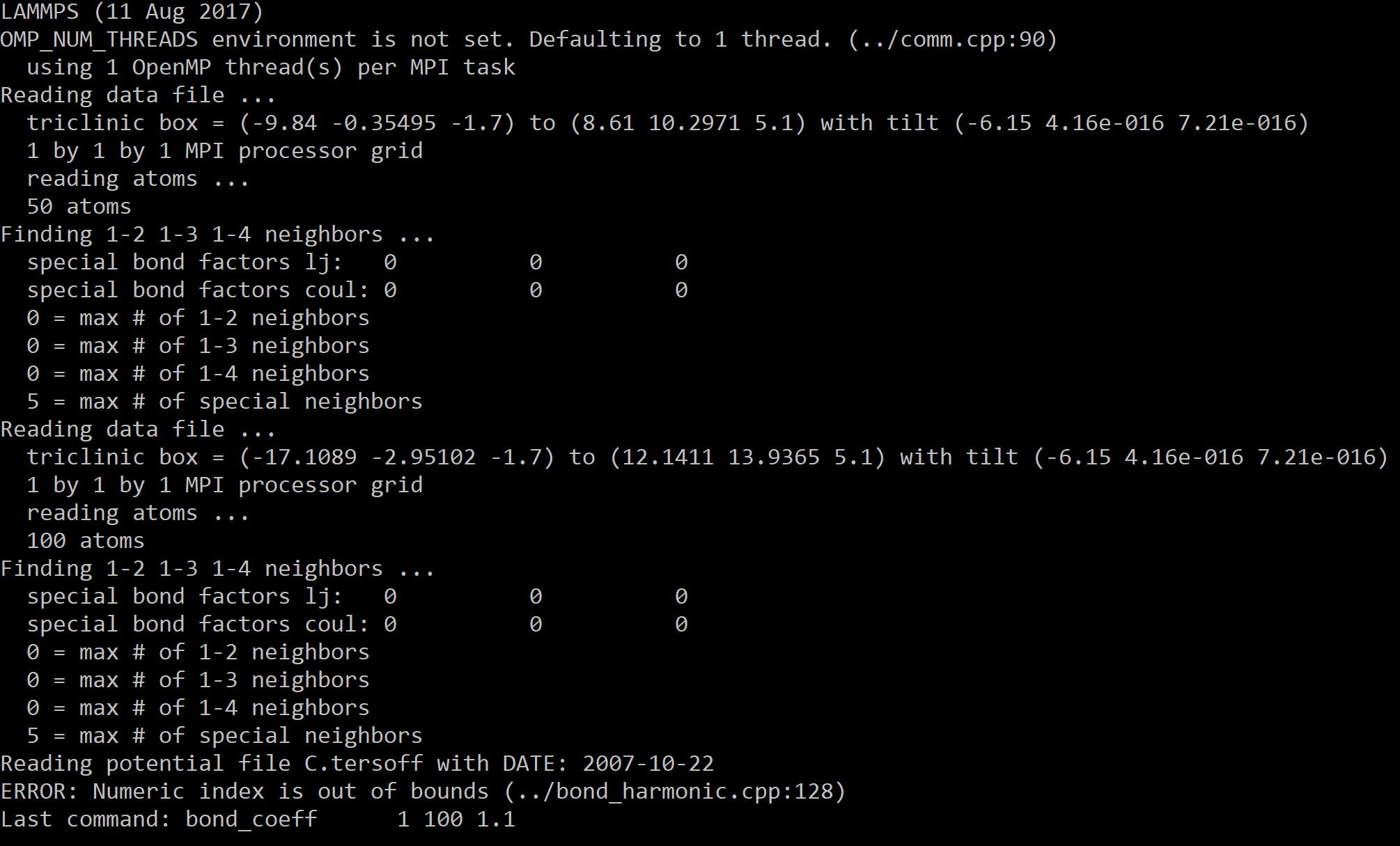
Hello,
Attached is my operation result with the error.
You are using a very, very old LAMMPS version.
I am attaching a minimal example to demonstrate that these flags work correctly with the current version of LAMMPS as can be seen from the output below.
LAMMPS (27 May 2021)
using 1 OpenMP thread(s) per MPI task
Reading data file …
orthogonal box = (-10.000000 -10.000000 -10.000000) to (10.000000 10.000000 10.000000)
1 by 1 by 1 MPI processor grid
reading atoms …
1 atoms
Finding 1-2 1-3 1-4 neighbors …
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
0 = max # of 1-2 neighbors
0 = max # of 1-3 neighbors
0 = max # of 1-4 neighbors
3 = max # of special neighbors
special bonds CPU = 0.000 seconds
read_data CPU = 0.003 seconds
Reading data file …
orthogonal box = (-10.000000 -10.000000 -10.000000) to (10.000000 10.000000 10.000000)
1 by 1 by 1 MPI processor grid
reading atoms …
1 atoms
Finding 1-2 1-3 1-4 neighbors …
special bond factors lj: 0 0 0
special bond factors coul: 0 0 0
0 = max # of 1-2 neighbors
0 = max # of 1-3 neighbors
0 = max # of 1-4 neighbors
3 = max # of special neighbors
special bonds CPU = 0.000 seconds
read_data CPU = 0.000 seconds
Neighbor list info …
update every 1 steps, delay 10 steps, check yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 5.3
ghost atom cutoff = 5.3
binsize = 2.65, bins = 8 8 8
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) pair zero, perpetual
attributes: half, newton on
pair build: half/bin/newton
stencil: half/bin/3d
bin: standard
(2) fix bond/create, occasional, copy from (1)
attributes: half, newton on
pair build: copy
stencil: none
bin: none
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 6.506 | 6.506 | 6.506 Mbytes
Step Temp E_pair E_mol TotEng Press
0 0 0 0 0 0
10 0 0 40.5 40.5 -0.015
Loop time of 6.4992e-05 on 1 procs for 10 steps with 2 atoms
System init for write_data …
Total wall time: 0:00:00
in.create (323 Bytes)
1.data (139 Bytes)
2.data (139 Bytes)
Hello,
Thanks for your help. I have downloaded a new LAMMPS version but the system automatically runs the old version. Should I uninstall the old version?
I cannot help you with that. What you need to do is dependent on your specific setup and how you installed the new version.
For the most part that is an issue of knowing how to use the operating system it is running and exactly what steps you did.
Thus it is both (mostly) off-topic for this mailing list and (nearly) impossible to advise on from remote.
Hello,
Thanks for your help, I have installed the new lamps version and successfully ran the example project you sent to me. However, when I run my own project, a new error occurred:
ERROR on proc 0: All pair coeffs are not set (src/pair_hybrid.cpp:682)
I did not change any code of my potential file, but this happened when I ran it with the new lamps version. Could you please give me some ideas why this happened?
Great thanks
Hello,
Thanks for your help, I have installed the new lamps version and successfully ran the example project you sent to me. However, when I run my own project, a new error occurred:
ERROR on proc 0: All pair coeffs are not set (src/pair_hybrid.cpp:682)
I did not change any code of my potential file, but this happened when I ran it with the new lamps version.
did not change anything compared to what?
please note that your original input file did not even reach this stage (i.e. the beginning of a “run”).
Could you please give me some ideas why this happened?
there have to be pair_coeff settings for all combinations of atom types. when you merge two systems, also for the interactions between the two systems.
this error message is an (obvious) indication that there are some combination of atom types for which no settings have been made.
Hello,
I have resolved the problem. The issue was that some pair atoms were not set with a potential. Thanks for your help, I am really appreciated.
Sincerely