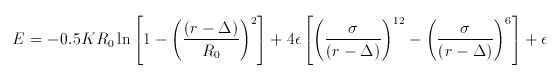I wrote a “fix_axial” in the spirit of “fix_uniaxial” but to independently control any of the 3 axes which is periodic (not necessarily conserve volume). The idea is that I want to control sigma_xx and epsilon_yy, so I would apply a fix_nph on x-axis to keep sigma_xx around zero and use my fix to expand/contract y-axis. (I live in 2D)
My “fix_axial” works perfectly fine by itself, but when I try to use it along with fix_nph to keep sigma_xx=0, the x-axis doesn’t seem to do any rescaling even when sigma_xx starts to increase.
Any gotchas anyone can think of? Looks like fix_nph should leave domain->boxylo, etc untouched and my fix_axial leaves domain->boxxlo, etc untouched.
I found this formula for fene/expand bond_style in Lammps' documentation. I
was wondering why this potential has a singularity at r=\delta. That seems to
mean the distance between two atoms are not allowed to be \delta.
I attached formula here.
Thank you.
Sorry, just ignore my message. I think I got it (It requires distances
between bonded atoms to be larger than \delta). The potential doesn't apply
to r <= \delta.
I tried a simulation with FENE potential. However harder I tried to tune
the parameter, it always prints hundreds of thousands of lines warning
message:
WARNING FENE bond too long ....
Basically I set a fene/expand potential within [0.4,1.0] ( R=0.6,
delta=0.4, sigma=0.26727 which will make LJ part in the FENE to have a
minimum at r=0.7). Also I set all pair interaction as LJ/expand ( delta=0.4,
sigma=0.26727).
Initially all bond lengths are 0.7 and a NVT simulation is run. Please
could anyone find what's wrong with my input?
Thank you very much.
For fene/expand, the delta = 0.0 is the same as fene.
For fene, two bonded atoms cannot be on top of each other (r = 0)
else the LJ term blows up. Ditto for fene/expand if the two
atoms are separated by delta.
"Bond too long" happens usually because a bond blew apart
b/c the atoms were forced too hard.
For the fene potential the equilibrium bond length is about 0.97 sigma
if sigma = 1.0 which is the sum of both terms, not just the LJ.
So for delta = 0.4, the equil would be about 1.37. So you need to
make sure your initial separations are not too small, else you will
blow apart the bonds.
Hi,
I am planning to do mesoscale simulation of a polymer system. From what i understood that the monomer or part of the monomer is represented as beads to do tha calculation. What i am little confused is 'if i chose a benzene ring to be a bead in a rod like polymer then during mesoscale dynamics how it takes care of the rotation of the ring around the axis of the rod. And this question also leads to the next one for the same reason i.e how to convert the beads to the atomistic model again.' I could not find these details as such anywhere and would appreciate any reference or inputs in this matter.
If you're representing an entire ring as a spherical bead, then
it has no orientation. At least beads in LAMMPS currently do not.
If you want some kind of spherical particle with orientation
then you would need to add that to LAMMPS as an atom
type and compute all the torques in a new pair potential
and integrate the eqs of motion for such an object in a new fix.