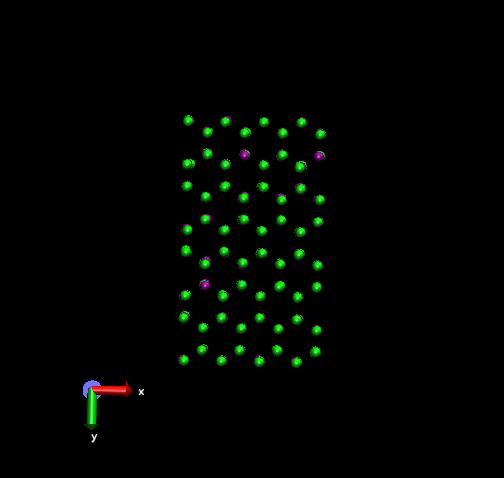
To calculate the formation energy of vacancies, I first equilibrated my system under NPT conditions and found the total energy. Then I removed 3 atoms of similar type and again found the energy of the system with these point defects. I removed 3 surface atoms so that it is easier for me to visualize the lattice with the removed atoms. The formation energy/atom (found by the difference in energies divided by 3) is similar to that in literature for a charge state of 0 for that type of vacancy.
2 atoms that were removed were surrounded by more atoms than the 3rd as shown in the attachment. So I thought of running the simulation with each of these atoms separately removed to see if there is a difference in formation energies. But the formation energy/atom are the same and similar to that found out earlier when all 3 atoms were removed simultaneously.
I just wanted to make sure if there is vacuum on all sides outside my specified 3D region or does LAMMPS assume that the region is part of the bulk i.e. automatically creates an infinite crystal with the region with 512 atoms (in my case) embedded within it? When I use the porosity command to randomly remove a fraction of the atoms, I see distortion of the lattice. Is this distortion just too little for me to observe when 3 atoms are removed? Or should I see a change in position of the underlying N atom to which the Ga atom was bonded? How do I track the atom ID for the atom sitting below the atom that I have removed?
Thanks,
Mihir.