Dear LAMMPS users:
I am modelling a MnO nanowire in a large box (with vacuum regions outside the side surfaces). I used to do it with DL-Poly software, it runs well. Now, with LAMMPS, I found the atoms was abnormally spreaded into the whole box (Please refer to the attached pic). Below is my input file, which I suspect something wrong with it. Any suggestions?
# MnO Nanowire, 28A thick, 6050 atoms
# Initialization
units real
atom_style atomic
boundary p p p
# Atom definition
read_data MnO.inp
###read_restart restart.MnO.*
# Setting
## Atomic group
group 1 type 1 3
group 2 type 2 4
## Forcefield
pair_style buck 6.0
pair_coeff 1 3 1007.4 0.3299 0
pair_coeff 1 4 1007.4 0.3299 0
pair_coeff 2 3 1007.4 0.3299 0
pair_coeff 2 4 1007.4 0.3299 0
pair_coeff 3 3 9547.96 0.21916 32
pair_coeff 3 4 9547.96 0.21916 32
pair_coeff 4 4 9547.96 0.21916 32
#zero
pair_coeff 1 1 0 0.2 0
pair_coeff 1 2 0 0.2 0
pair_coeff 2 2 0 0.2 0
## Parameter of simulation
neighbor 0.3 bin
neigh_modify every 50 delay 0 check no
timestep 2.0
fix mobile 2 nvt 300. 300. 100.
#Set frozen atoms
fix freeze_atoms 1 setforce 0.0 0.0 0.0
# Output
dump mydump all custom 2000 dump.MnO tag type x y z vx vy vz fx fy fz
thermo 50
restart 2000 restart.MnO
See all the ways you can stay connected to friends and family
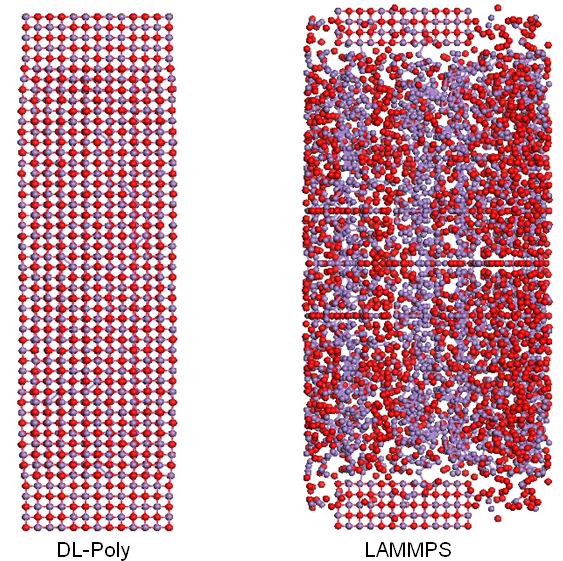
You are using periodic BC, is that what you want to
have large vacuum around the nanowire? Is the time 0
snapshot correct? Are you saying the dynamics don't do
what you expect?
Steve
Hi, Steve:
My periodic cell is much larger than the region that atoms occupying, i.e. the model has actually free surfaces. At time 0, the model snapshot is correct. While start running equilibration (at 300K), the atoms move much faster than expected. I have suspected it comes from the "real’ unit of buck potential parameter, but it was not after testing.
Thanks for attention.
Scott
BTW: In my output, the Temperature & total energy has intensive fluctuations, is it reasonable?
Step Temp E_pair E_mol TotEng Press
0 0 19830.779 0 19830.779 11168.923
50 162.70043 18059.933 0 20993.571 10746.144
100 755.97883 14752.631 0 28383.624 10745.502
150 173.04959 13083.876 0 16204.119 8063.2405
200 65.422941 12161.927 0 13341.562 7257.2075
250 129.55207 10861.28 0 13197.223 6786.4646
300 416.08859 8429.5397 0 15932 6378.9563
350 337.36139 6240.538 0 12323.474 4948.9189
400 131.11734 5269.7249 0 7633.8911 3817.2113
450 114.08503 4765.2949 0 6822.3529 3473.6096
500 230.72648 4503.9427 0 8664.1534 3589.774
550 427.54341 4541.5857 0 12250.587 4170.3131
600 255.24147 4504.9192 0 9107.1577 3593.2724
650 144.20661 4114.9421 0 6715.1199 3027.8962
700 165.46614 3893.0143 0 6876.5209 2944.0285
750 306.27761 3930.24 0 9452.707 3366.4022
800 362.28754 3969.7311 0 10502.109 3532.1154
850 204.95214 3846.249 0 7541.7247 2980.859
900 151.59269 3720.4264 0 6453.782 2733.5737
950 217.72131 3690.5553 0 7616.2709 2907.8835
1000 367.08542 3698.9817 0 10317.87 3352.6022
1050 288.52055 3640.8653 0 8843.1562 3075.3366
1100 164.86524 3660.0893 0 6632.7613 2700.6485
1150 162.12846 3779.5034 0 6702.8287 2744.562
1200 308.28262 3591.9281 0 9150.5473 3071.4517
1250 350.13194 3821.8839 0 10135.085 3339.5653
1300 201.48779 3854.1654 0 7487.1756 2895.0938
1350 158.55298 3759.5648 0 6618.4209 2705.9053
1400 233.31747 3684.9643 0 7891.8929 2899.7723
1450 358.81586 3562.9965 0 10032.777 3195.3923
1500 261.45002 3631.6302 0 8345.8145 2946.7847
1550 170.38293 3552.6122 0 6624.7732 2633.8099
1600 188.50378 3520.0773 0 6918.9738 2675.7106
1650 304.94765 3532.4876 0 9030.9743 3035.6769
1700 320.82927 3667.7804 0 9452.6273 3176.0684
1750 206.14307 3680.4146 0 7397.3637 2837.9636
1800 175.66095 3589.5689 0 6756.8974 2694.5818
1850 238.39801 3652.4432 0 7950.9786 2925.9876
1900 328.77881 3798.9188 0 9727.1033 3292.2591
1950 266.07328 3877.3268 0 8674.8728 3146.3374
2000 188.61071 3829.7894 0 7230.6141 2882.5612
You are only reneighboring every 50 steps which could
be a problem. I would neighbor every step with check = yes,
so the code will only do it when needed. Your T fluctuations
do seem large, assuming the initial sample is reasonably well
equilibrated.
Steve
Hi, Steve:
Thanks for your prompt reply. After trying, it seems reneighboring doesn't help.
I am now suspecting the coefficient setting of Buck potential, as the pair-coefficient from input & output (from restart.xxx file & converted with restart2data) doesn't show the same (as below). Does it go wrong?
input:
## Atom type 1,2 for Mn atoms & 3,4 for O atoms
## Forcefield
pair_style buck 6.0
pair_coeff 1 3 23230.644 0.3299 0 # Mn-O interaction
pair_coeff 1 4 23230.644 0.3299 0
pair_coeff 2 3 23230.644 0.3299 0
pair_coeff 2 4 23230.644 0.3299 0
pair_coeff 3 3 220175.958 0.21916 737.92 # O-O interaction
pair_coeff 3 4 220175.958 0.21916 737.92
pair_coeff 4 4 220175.958 0.21916 737.92
#zero
pair_coeff 1 1 0 0.2 0 # Mn-Mn interaction
pair_coeff 1 2 0 0.2 0
pair_coeff 2 2 0 0.2 0
Output (from restart.**):
LAMMPS data file from restart file:
................
Pair Coeffs
1 0 0.2 0
2 0 0.2 0
3 220176 0.21916 737.92 # only O-O interactions??
4 220176 0.21916 737.92
Rgds!
Scott
Date: Tue, 21 Jul 2009 07:57:29 -0600
Subject: Re: [lammps-users] FW: Problem: Atomic position out-of-control
From: [email protected]
To: [email protected]...
CC: [email protected]
You are only reneighboring every 50 steps which could
be a problem. I would neighbor every step with check = yes,
so the code will only do it when needed. Your T fluctuations
do seem large, assuming the initial sample is reasonably well
equilibrated.
Steve
> Hi, Steve:
>
> My periodic cell is much larger than the region that atoms occupying, i.e.
> the model has actually free surfaces. At time 0, the model snapshot is
> correct. While start running equilibration (at 300K), the atoms move much
> faster than expected. I have suspected it comes from the "real' unit of
> buck potential parameter, but it was not after testing.
>
> Thanks for attention.
> Scott
>
>> Date: Fri, 17 Jul 2009 08:42:51 -0600
>> Subject: Re: [lammps-users] FW: Problem: Atomic position out-of-control
>> From: [email protected]
>> To: [email protected]...
>> CC: [email protected]
>>
>> You are using periodic BC, is that what you want to
>> have large vacuum around the nanowire? Is the time 0
>> snapshot correct? Are you saying the dynamics don't do
>> what you expect?
>>
>> Steve
>>
>> >
>> >
>> > ________________________________
>> > From: [email protected]...
>> > To: [email protected]...
>> > Subject: Problem: Atomic position out-of-control
>> > Date: Fri, 17 Jul 2009 13:48:31 +0800
>> >
>> > Dear LAMMPS users:
>> >
>> > I am modelling a MnO nanowire in a large box (with vacuum
>> > regions outside
>> > the side surfaces). I used to do it with DL-Poly software, it runs well.
>> > Now, with LAMMPS, I found the atoms was abnormally spreaded into the
>> > whole
>> > box (Please refer to the attached pic). Below is my input file, which I
>> > suspect something wrong with it. Any suggestions?
>> >
>> > # MnO Nanowire, 28A thick, 6050 atoms
>> >
>> > # Initialization
>> > units real
>> > atom_style atomic
>> > boundary p p p
>> >
>> > # Atom definition
>> > read_data MnO.inp
>> >
>> > ###read_restart restart.MnO.*
>> >
>> > # Setting
>> > ## Atomic group
>> > group 1 type 1 3
>> > group 2 type 2 4
>> > ## Forcefield
>> > pair_style buck 6.0
>> > pair_coeff 1 3 1007.4 0.3299 0
>> > pair_coeff 1 4 1007.4 0.3299 0
>> > pair_coeff 2 3 1007.4 0.3299 0
>> > pair_coeff 2 4 1007.4 0.3299 0
>> >
>> > pair_coeff 3 3 9547.96 0.21916 32
>> > pair_coeff 3 4 9547.96 0.21916 32
>> > pair_coeff 4 4 9547.96 0.21916
>> > 32
>> >
>> > #zero
>> > pair_coeff 1 1 0 0.2 0
>> > pair_coeff 1 2 0 0.2 0
>> > pair_coeff 2 2 0 0.2 0
>> >
>> >
>> >
>> > ## Parameter of simulation
>> > neighbor 0.3 bin
>> > neigh_modify every 50 delay 0 check no
>> >
>> > timestep 2.0
>> >
>> > fix mobile 2 nvt 300. 300. 100.
>> >
>> > #Set frozen atoms
>> > fix freeze_atoms 1 setforce 0.0 0.0 0.0
>> >
>> > # Output
>> > dump mydump all custom 2000 dump.MnO tag type x y z vx vy vz
>> > fx
>> > fy fz
>> > thermo 50
>> > restart 2000 restart.MnO
>> >
>> >
>> > ________________________________
>> > See all the ways you can stay connected to friends and family
>> > ________________________________
>> > Windows Live™: Keep your life in sync. Check it out!
>> >
>> > ------------------------------------------------------------------------------
>> > Enter the BlackBerry Developer Challenge
>> > This is your chance to win up to $100,000 in prizes! For a limited time,
>> > vendors submitting new applications to BlackBerry App World(TM) will
>> > have
>> > the opportunity to enter the BlackBerry Developer Challenge. See full
>> > prize
>> > details at: http://p.sf.net/sfu/Challenge
>> > _______________________________________________
>> > lammps-users mailing list
>> > [email protected]
>> > https://lists.sourceforge.net/lists/listinfo/lammps-users
>> >
>> >
>
> ________________________________
> check out the rest of the Windows Live™. More than mail–Windows Live™ goes
> way beyond your inbox. More than messages
With Windows Live, you can organize, edit, and share your photos.