Hi all,
I am currently trying to generate random configurations of spheres (atoms) to use as initial condition in an other code (CFD). I use the 3 Mar 2020 stable version.
To that purpose, I use the create atoms command with random option, followed by a granular simulation to separate the initially overlapping atoms.
Here is my input script :
units lj
atom_style sphere
boundary p p p
newton off
comm_modify vel yes
region reg block 0 12 0 12 1 40 units box
create_box 1 reg
create_atoms 1 random 2304 36908 reg
neighbor 0.5 bin
neigh_modify delay 0 every 1 check yes
pair_style gran/hooke/history 5000.0 NULL 50 NULL 0.5 1
pair_coeff * *
timestep 0.0002
fix 1 all nve/limit 0.1
compute 1 all erotate/sphere
thermo_style custom step atoms ke c_1 vol
thermo 500
thermo_modify lost warn norm no
compute_modify thermo_temp dynamic/dof yes
dump 1 all custom 10000 dump.random id x y z diameter
dump_modify 1 format line “%d %1.12e %1.12e %1.12e %1.12e”
dump_modify 1 sort id
dump 2 all image 10000 frames/rdm2304.*.jpg type type &
axes yes 0.8 0.02 view 80 -30
dump_modify 2 pad 6
run 200000
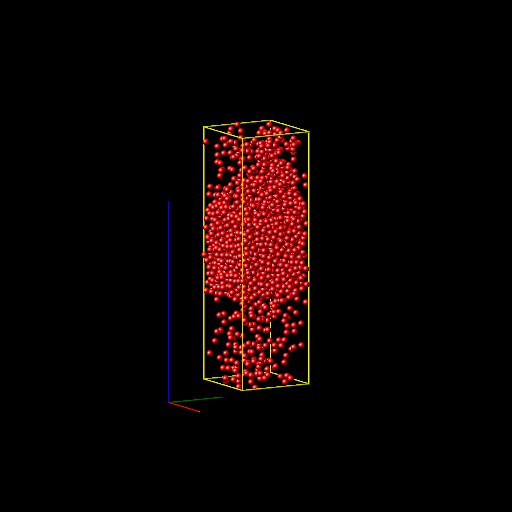
The script runs smoothly. However, I am surprised by the final result. The configuration obtained at the end of the granular simulation shows a strong aggregate of atoms in the middle of the vertical dimension (see attached file), although there is no gravity involved and the vertical direction is periodic just as the two transverse directions.
I have tried to reduce the time step or the nve/limit parameter, without success.
I would very much appreciate if any of you already ran into that kind of problem/unexpected outcome and found a solution.
Best,
B. Hardy
![]()