Dear Sandia,
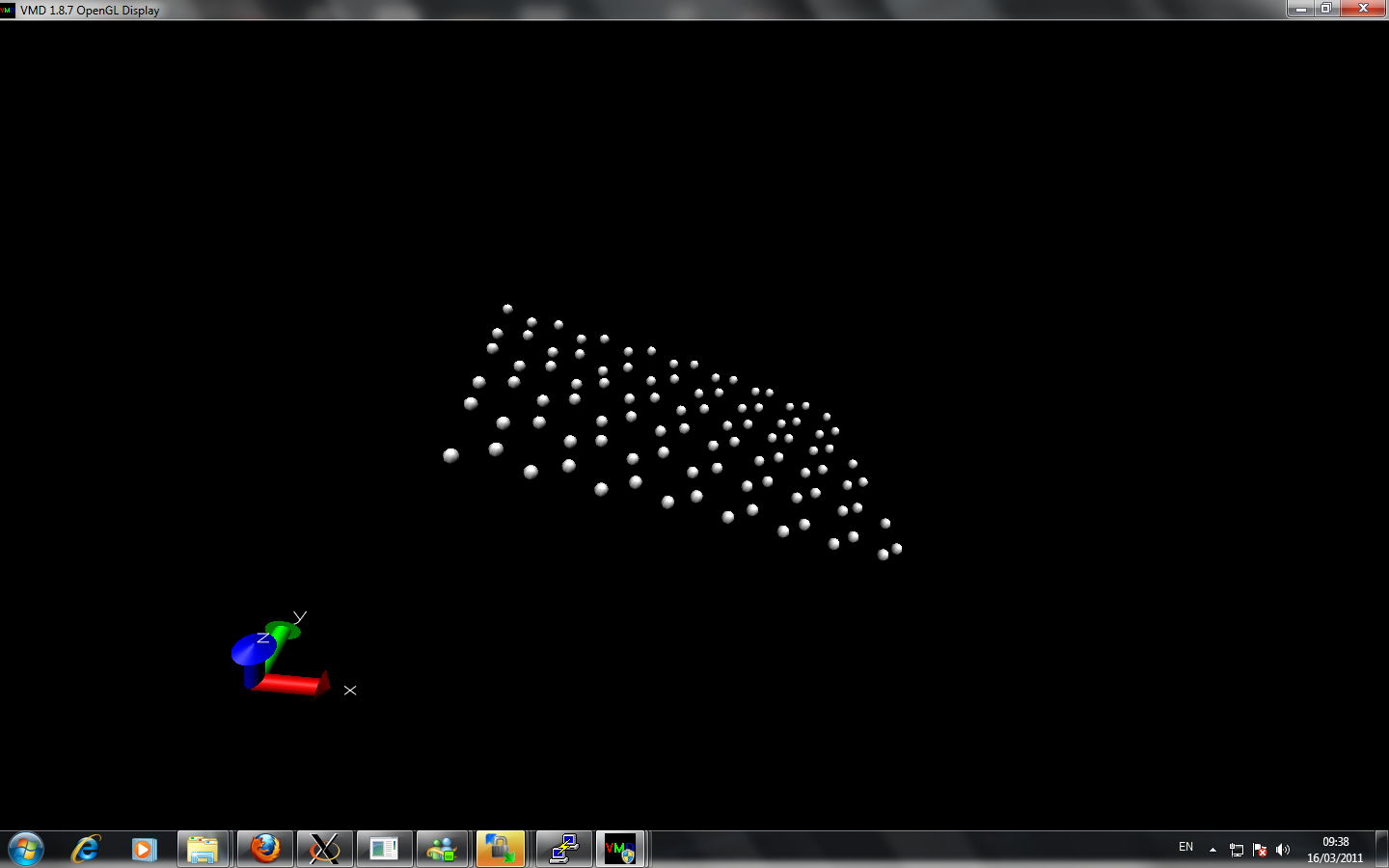
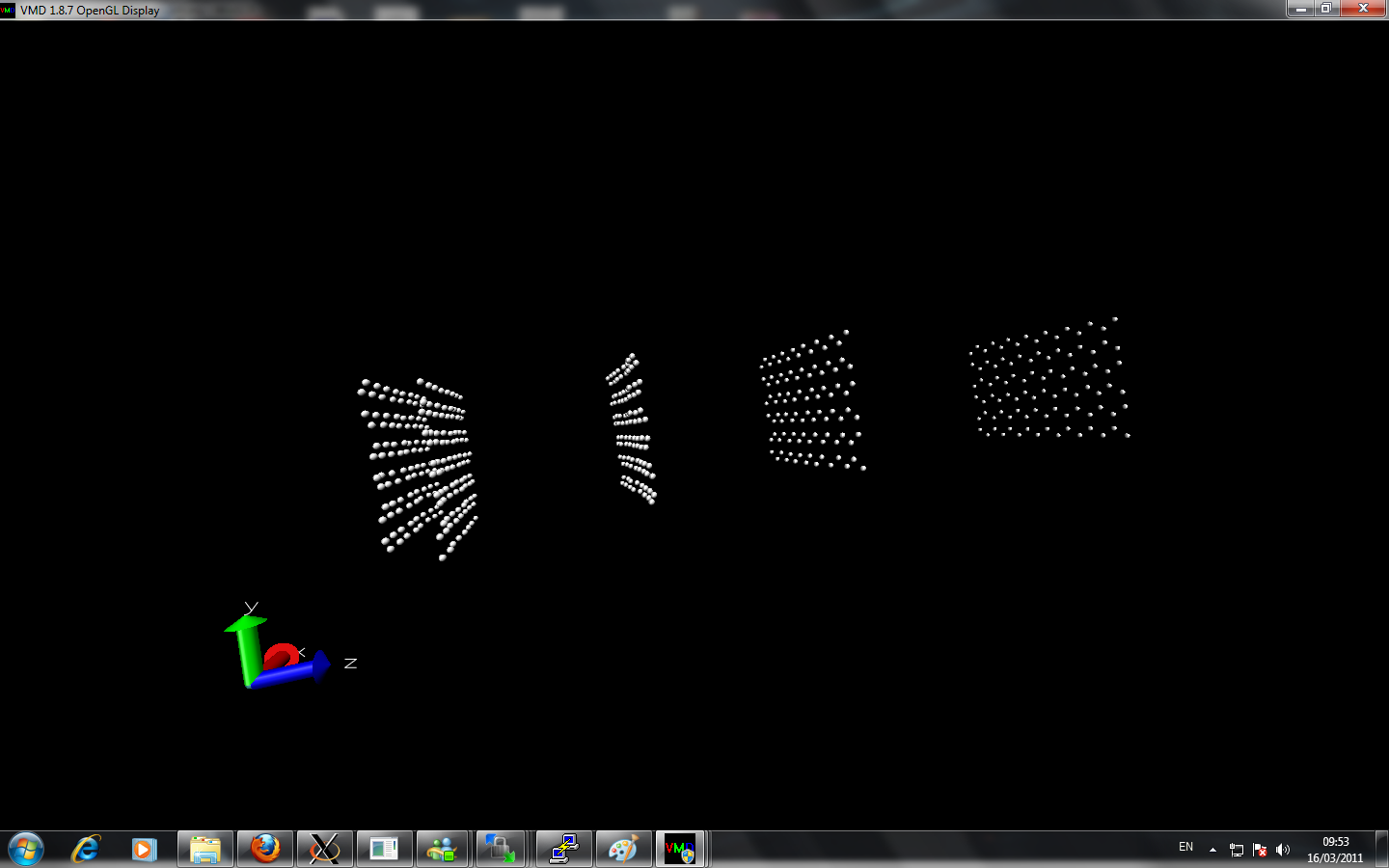
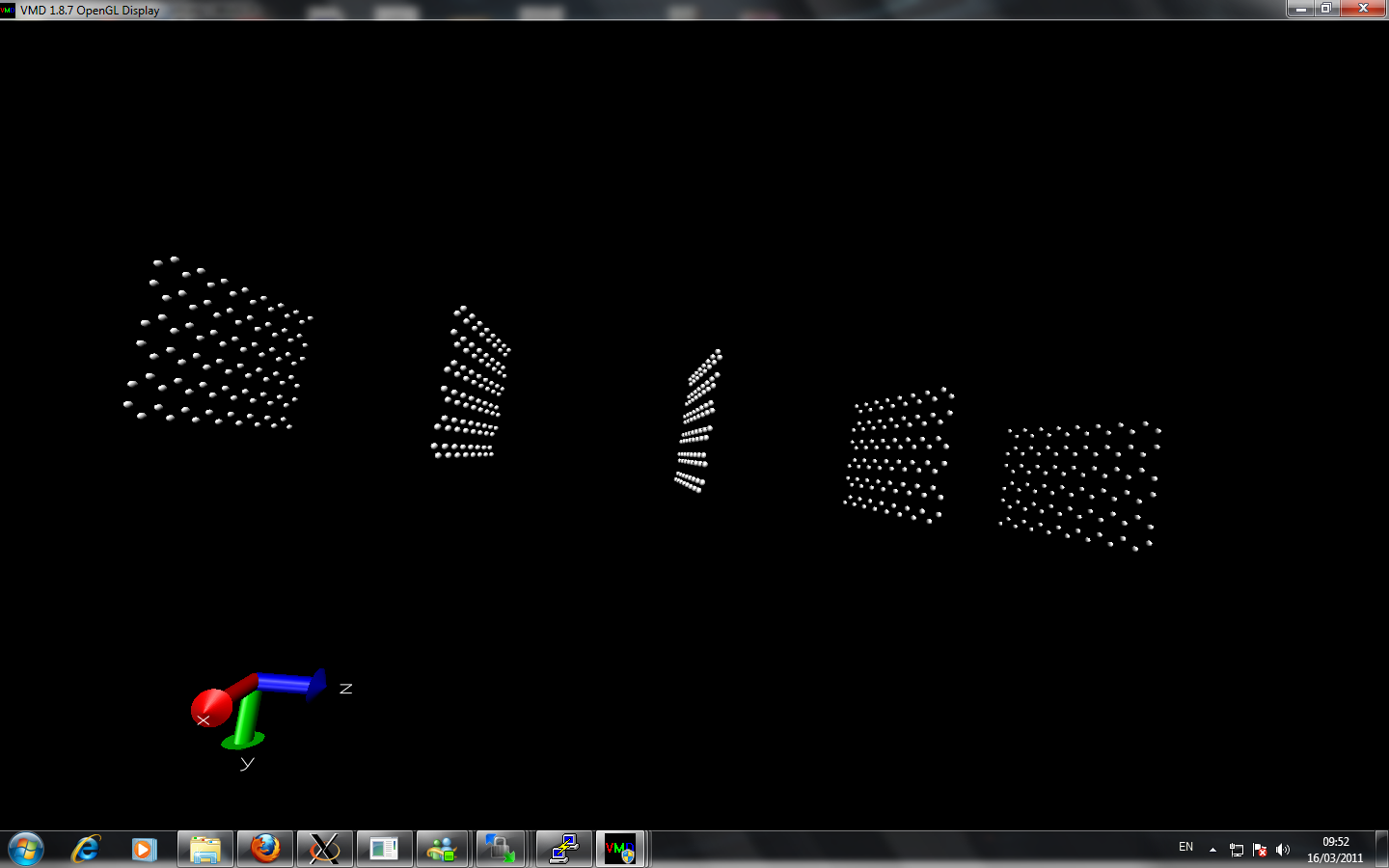
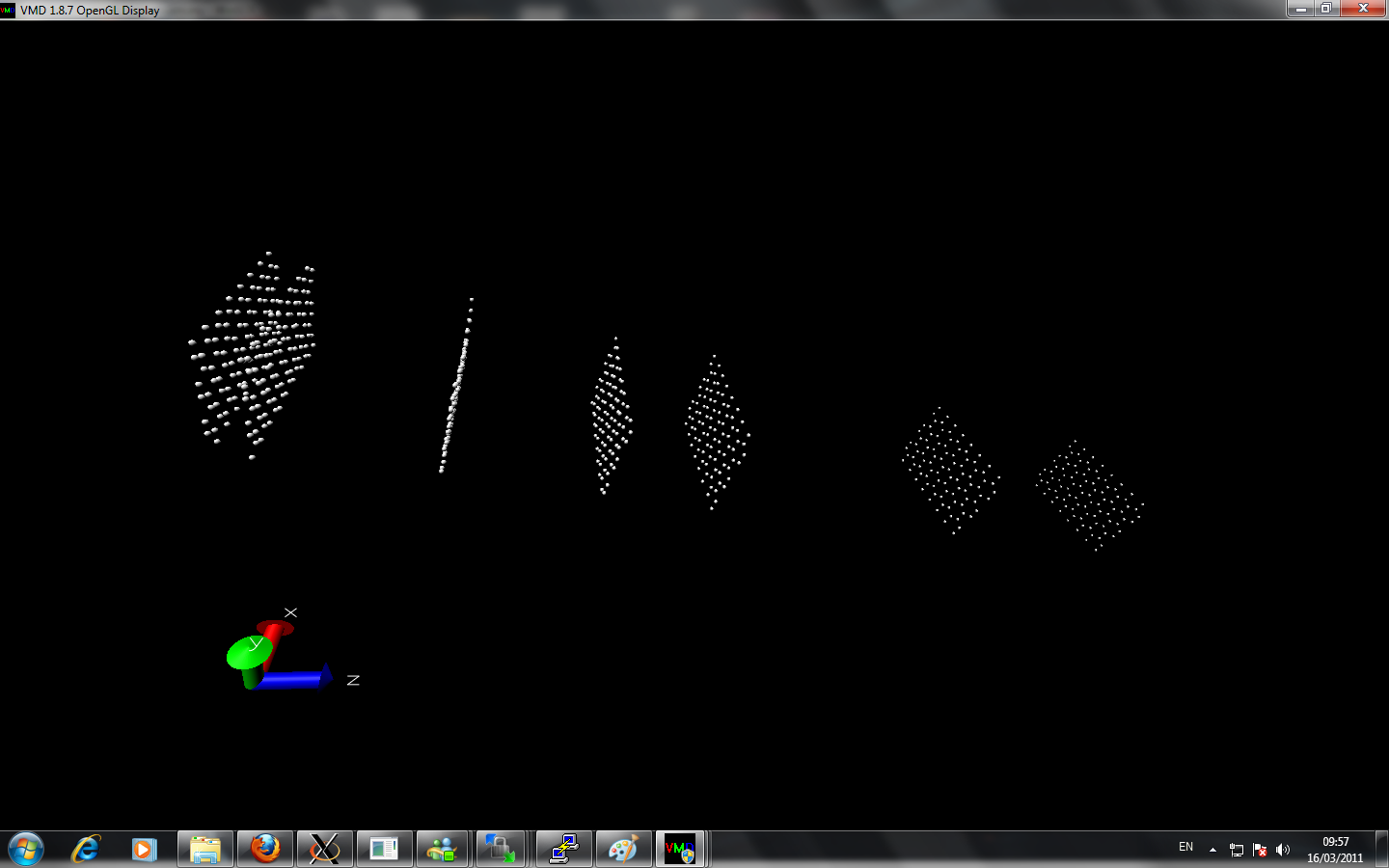
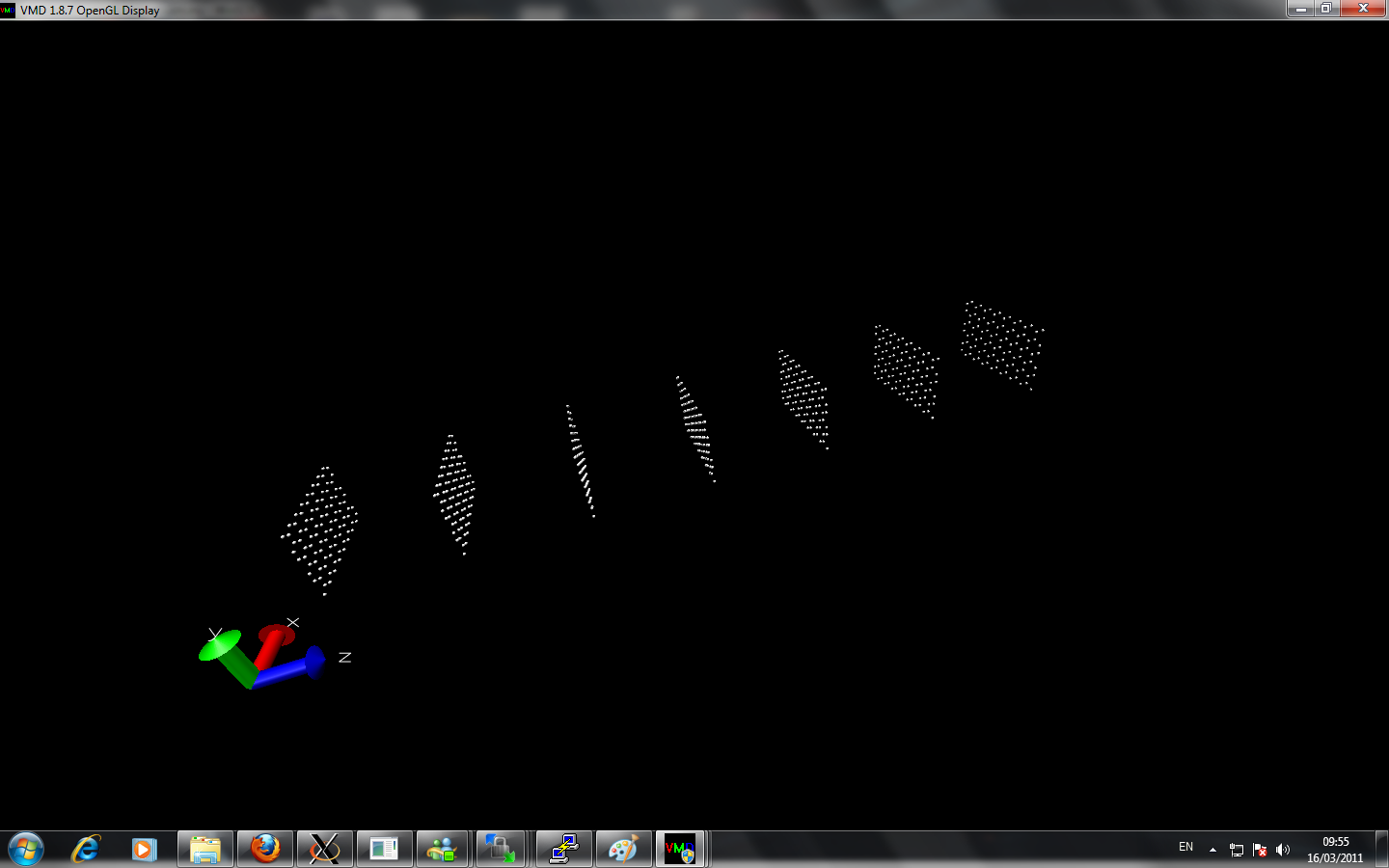
I am currently trying to calculate the thermal conductivity of different layers of graphene. I created multiple layer by using replicate command. However, when I perform two layers with nvt command at room temperature about 2 million timesteps, the two sheets from initially seperated far away come close together at equilibrate position. When I perform more layer graphene sheets, the graphene alone z-axis lost it's symmetry and at the beginning of z-axis the sheet seems are very close to each other while at the end of z-axis the seperation of the graphene via replicate is remained.
Can anyone point out there is any error of my simulation?
Here is the input script and image I attached:
dimension 3
boundary p p p
units metal
atom_style charge
read_data data.charge
region up block -9.838 -7.8704 INF INF INF INF units box
region down block 0 1.9676 INF INF INF INF units box
pair_style airebo 3.0 1 1
pair_coeff * * CH.airebo C
replicate 1 1 5
neighbor 4.0 bin
neigh_modify delay 0 every 1 check yes
timestep 0.00005
velocity all create 300 1234567 mom yes rot yes dist gaussian units box
fix thermostat all nvt temp 300.0 300.0 0.1
compute alltemp all temp
thermo_style custom step atoms temp press pe ke etotal xlo xhi ylo yhi zlo zhi vol enthalpy
thermo_modify temp alltemp lost warn
compute ke all ke/atom
variable temp atom c_ke/(1.5*1.0)/8.617343*100000.0
fix temp_profile all ave/spatial 1 100000 100000 x -9.838 1.9676 v_temp file temp6.profile units box
fix temp_atom all ave/atom 1 100000 100000 v_temp
compute up_temp all temp/region up
compute down_temp all temp/region down
variable delta_t equal c_down_temp-c_up_temp
fix delta_t all ave/time 1 100000 100000 v_delta_t file delta_t6.dat
dump 1 all xyz 100000 dump6.coord.*
dump 2 all custom 100000 dump6.vel.* id type mass vx vy vz fx fy fz f_temp_atom
restart 400000 restart6.mwnt.*
thermo 1000
run 400000
unfix thermostat
fix ensemble all nve
fix heat_swap all thermal/conductivity 40 x 10
fix e_exchange all ave/time 40 2500 100000 f_heat_swap file e_exchange6.dat
run 2000000