Dear LAMMPS developers and users,
I am trying to compute the phonon density of states of graphene with LAMMPS. But in my result, the intensity is very large when frequency is around 0Hz, which is inconsistent with the usual result. My computation method is listed as follows.
- Dump velocities(Vx, Vy and Vz) of each atoms in a certain region every N(=5) time steps to a file with LAMMPS
- For each atom, calculate its auto-correlation function.
- Conduct FFT to the auto-correlation functions. That is the DOS of that atom.
4 Average the results of all atoms.
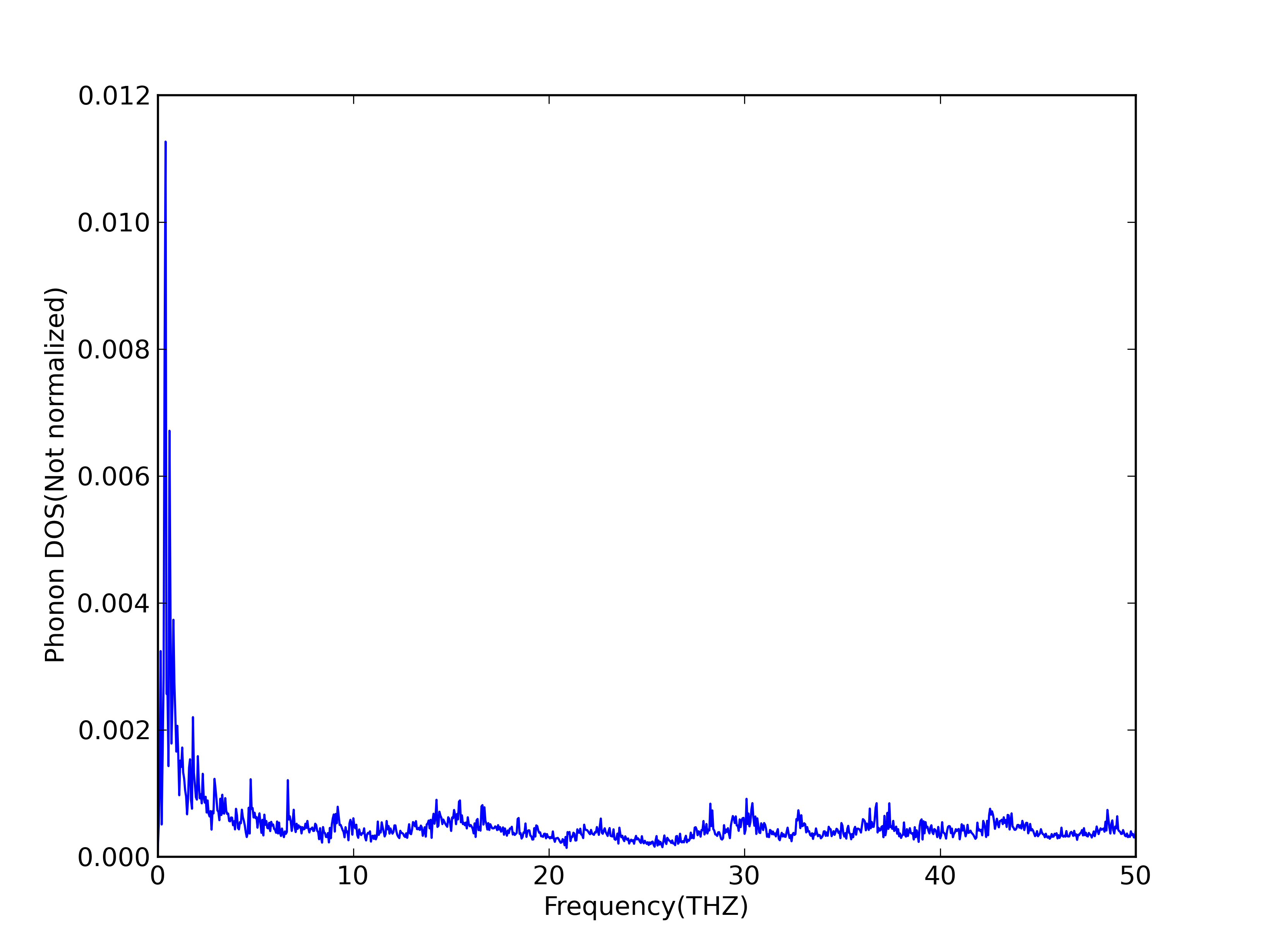
But my result seems to be wield(please see the attachment of this email). The intensity near zero frequency is very large, but it is usually zero in the papers I read before. Could you shed some light on this for me?
Thanks,
Yan
Dear LAMMPS developers and users,
I am trying to compute the phonon density of states of graphene with LAMMPS.
But in my result, the intensity is very large when frequency is around 0Hz,
which is inconsistent with the usual result. My computation method is listed
as follows.
1. Dump velocities(Vx, Vy and Vz) of each atoms in a certain region every
N(=5) time steps to a file with LAMMPS
2. For each atom, calculate its auto-correlation function.
3. Conduct FFT to the auto-correlation functions. That is the DOS of that
atom.
4 Average the results of all atoms.
But my result seems to be wield(please see the attachment of this email).
The intensity near zero frequency is very large, but it is usually zero in
the papers I read before. Could you shed some light on this for me?
it is really difficult to tell what went wrong, but my first guess would be
that you mixed up velocities from different atoms.
have you sorted the velocities by atom id before postprocessing?
have you tried computing the auto-correlation from within lammps instead?
cheers,
axel.