Dear lammps users and developers, I had a problem regarding simulating a
lipid membrane subjected to water flow .
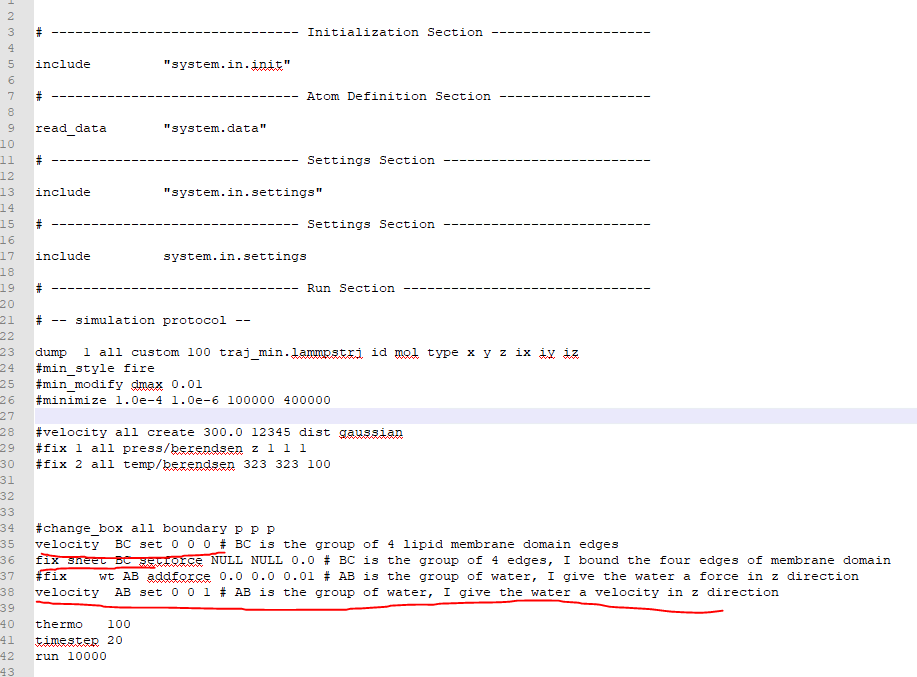
The coarse lipid membrane configuration is positioned in the x-y domain
with a rectangular (with a thickness) setting with 4 edges . The water
molecule is distributed above and below the membrane. I tried to study the
water flow effects on the membrane. So I bounded the 4 edges of the
membrane and give water a velocity in the z-direction as shown below,
[image: image.png]
But the simulation results show that the membrane and water are still
static. So could any suggestions be given in terms of this simulation?
Thanks for your attention in advance.
Best,
difficult to say what is going on without seeing all of your input files. there is no time integration fix shown here, so that could be a reason.
it is also odd that you include the “system.in.settings” file twice.
axel.
Thanks for the quick feedback, I think that is the reason but I don't
realize it until your guide, could you give a link on how to set the time
integration if you are convenient? I really appreciate any suggestions
from your side. Thanks again!
Best,
My recommendation is 3-fold.
- please study the LAMMPS manual and some LAMMPS tutorials. for example the one hosted at https://tutorial.lammps.org.
- please do not start simulations of complex systems (like the one you are planning to do) without having first gained practice simulating simpler systems (e.g. lipidmembrane only, water only, water with flow only). also when starting the final simulation, you should build the input and setup in stages (e.g. equilibrate a static system before inducing a flow).
- discuss with your adviser/tutor how to properly set up and run meaningful simulations. while I can give you a command line that will overcome the immediate issue, your simulation will likely be bogus and for something as fundamental as this, you should be knowing yourself why and how to do this. please keep in mind that performing MD simulations is as much a craft as a science, so you have to approach it as such. having the tools (software and commands) does not guarantee that you get something that is correct and meaningful. that often requires a certain degree of practice, experience, and insight.
please also keep in mind that most of this is not something that you can be taught on a mailing list and thus it is considered (mostly) off-topic.
axel.
Yes, you are correct, Axel, Sorry for any inconvenience I brought in terms
of my issues,
I am just a beginner in this area and my supervisor or tutor... even
doesn't know MD (of course that is my problem), this mail list is the only
source I can get any more accurate information from during the pandemic.
before I inquire about this problem I know there must be some setting lost
but I just do not know what that is.
your detailed suggestions are really helpful in directing me in this field.
Thanks for your help.
Best,
*Wenhao Yao*
*First-Year Ph.D. Student*
*Theoretical and Applied Mechanics Laboratory*
*Department of Mechanical Engineering University of Alberta*
*4-225 Donadeo Innovation Centre for Engineering*
*9211 - 116 Street NW*
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
Edmonton, Alberta, Canada
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
T6G
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
*1H9*
*T. +1.587.938.7676*
*E. [email protected] <[email protected]>*
Thanks for the quick feedback, I think that is the reason but I don’t realize it until your guide, could you give a link on how to set the time integration if you are convenient? I really appreciate any suggestions from your side. Thanks again!
Best,
Wenhao Yao
First-Year Ph.D. Student
Theoretical and Applied Mechanics Laboratory
Department of Mechanical Engineering
University of Alberta
4-225 Donadeo Innovation Centre for Engineering
9211 - 116 Street NW
Edmonton, Alberta, Canada T6G 1H9
T. +1.587.938.7676
E. wyao1@ualberta.ca
Yes, you are correct, Axel, Sorry for any inconvenience I brought in terms of my issues,
I am just a beginner in this area and my supervisor or tutor… even doesn’t know MD (of course that is my problem),
i consider it a very, very bad idea to start doing MD simulations (and such complex ones to boot) without proper guidance or supervision.
you are bound to make all them many mistakes that beginners in MD do. there is a significant amount of “residual knowledge” in groups that do MD simulations that you are missing.
I am very certain that there are people with such knowledge at your institution and if you are (and your supervisor) are serious about getting into MD simulations you should reach out to one of them and start a collaboration/cooperation what would start with getting the proper tutoring.
this mail list is the only source I can get any more accurate information from during the pandemic. before I inquire about this problem I know there must be some setting lost but I just do not know what that is.
there are going to be more problems (just add the line “fix 0 all nve”) to your input and you will see. please note that MD simulations follow the “garbage in, garbage out” principle, that is you can only get meaningful results, if everything in the input is meaningful and properly done. you can have simulations that finish without crash and look like they were working correctly and still be completely bogus. this is further complicated by the property that what is a good choice in one context, would be a disaster in another.
axel.
Yeah, I had the command "fix 0 all nve" previously that I guess might be
missing. The results are just you have mentioned......you can imagine.
Anyhow, I will consider your words seriously, your work during the mailing
list really help a lot for lammps users at all levels.
Best,
*Wenhao Yao*
*First-Year Ph.D. Student*
*Theoretical and Applied Mechanics Laboratory*
*Department of Mechanical Engineering University of Alberta*
*4-225 Donadeo Innovation Centre for Engineering*
*9211 - 116 Street NW*
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
Edmonton, Alberta, Canada
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
T6G
<https://maps.google.com/?q=9211+-+116+Street+NW+Edmonton,+Alberta,+Canada++T6G&entry=gmail&source=g>
*1H9*
*T. +1.587.938.7676*
*E. [email protected] <[email protected]>*
Hi Wenhao
I agree with Axel that this is a relatively complicated simulation. It is possible to do, but there are a lot of issues. I have some deadlines now and cannot provide you with the detailed in-depth help that you would benefit from. But I’ll do the best I can in a short time this evening.
From communication I have had with you privately, I have a pretty good idea what you are trying to simulate. (The corresponding moltemplate example is here, and there is a picture of it at t=0 here.)
It seems like you want to immobilize the lipids at the edges of the simulation box and apply a pressure in the +Z direction that makes the membrane bulge upwards. To do this, you are attempting to force the water to move at constant velocity in the Z direction. The “velocity” command you are using only sets the velocity at the beginning of the simulation. The membrane blocks the atoms, so they will stop moving when they bump into it. That’s probably why it appears like nothing is moving. There is no way for the water to flow in the Z direction. The membrane is blocking the water.
-
Instead, why don’t you apply a force to the water particles in the +Z direction? This would establish a pressure difference and make the membrane bulge upwards. I assume that is what you want. (It looks like you tried this but you commented it out.)
-
Avoid using the Berendsen thermostat algorithm. It is a very old method that causes many serious problems. (Personally, I think the developers should remove the Berendsen thermostat from LAMMPS.)
Instead, for simulations of this type, I like to use a combination of both “fix langevin” and “fix nph”. But you can also use “fix npt” if you prefer (as a replacement for “langevin”+“nph”). But please avoid the Berendsen thermostat.
-
You are using the “iso” keyword with the pressure barostat. This is a mistake. You are simulating a system which is not symmetric in X,Y,Z. So you must allow the size of the simulation box in the X,Y,Z directions to equilibrate independently. Otherwise you might crush the membrane while attempting to relax the pressure in the water (or visa-versa). The “run.in.npt” file that comes with the moltemplate example I mentioned above, provides an example how to do this.
-
You must wait for the lipid density in the membrane to equilibrate before you begin immobilizing the lipids at the 4 edges of the boundary box. If you initially prevent these lipids from moving there will be no way to relax the surface tension in the membrane. Hence the size of the simulation box in the XY direction will remain the same.
You could run a short simulation under NPT conditions using a non-isotropic barostat to do this. (Long enough for the lipid density to equilibrate, but short enough so that the lipids at the edge do not move very far away from their initial positions. Example picture here.) Then you can immobilize the lipids. (See below for suggestions for how to do that.) You will have to determine how long to run this simulation by trial and error.
Alternatively if the lipids in the group that you wanted to immobilize have moved too far while equilibrating the membrane tension, you can use the “region” and “group” commands to create a new group of atoms that lie on the edges of the periodic boundaries and immobilize them. This is probably the best approach, but I have never tried this so you will have to figure this out. (See the links I provided.) (Also, because of periodic boundary conditions, it is enough to immobilize the particles near the XZ and YZ planes. You don’t need to immobilize all 4 sides of the square.)
-
You want to immobilize the particles in your simulation (the lipids on the sides of the periodic boundary box). Do not use “fix setforce” to do this.
Simulating an immobile object under NVT conditions can be done by excluding those atoms from the group of atoms that you send to the fixes that integrate the equations of motion (“fix langevin” and “fix nve”, or “fix nvt”).
Simulating a NPT conditions is much more difficult. My best attempts to do this are outlined in the “run.in.npt” files which are available in these two moltemplate examples.
a) translocation example (run.in.npt file here)
b) nanotube+water example (run.in.npt file here)
In these examples, I was using “fix rigid” on the immobile particles in an effort to correct the virial and stabilize the pressure. But this method is controversial. (There was a long discussion of this topic here.) For this reason, I would equilibrate the pressure first, and then immobilize the particles when running the simulation at NVT conditions.
---- summary ----
Here is what I would do.
-
Don’t use “velocity” or “fix move” or “fix momentum” or “fix setforce” to immobilize the lipids or make the water move upwards and avoid all of the berendsen fixes.
-
Wait for the pressure of the system to equilibrate first (using a combination of “fix nph” and “fix langevin”, or “fix npt”, together with the “aniso” keyword).
-
Then define the group of atoms on the edge of the periodic boundaries that you want to immobilize (using the “region” and “group” commands).
-
Then use the “group” command again to define the group of atoms that you want to move freely. If the group of atoms you want to immobilize is called “gImmobile”, you can define the group of mobile atoms (“gMobile”) this way:
group gMobile subtract all gImmobile
-
Use this group (eg. “gMobile”) with the fixes that integrate the equations of motion under NVT conditions (ie. “fix langevin” and “fix nve”, or “fix nvt”).
-
Use “fix addforce” to add a force to the water pushing them in the +Z direction.
-
My most important suggestion:
At the beginning, do not try to create a simulation that does all of these things.
Try running a simple simulation first. Then gradually and complexity. If you try to build a simulation that does all of these things it will probably fail, and it will be difficult to figure out what went wrong. Start with a simple simulation of a lipid in water under NPT anisotropic conditions. (As in the moltemplate example.) Then gradually add complexity, one fix at a time.
I hope that gets you started.
Cheers
Andrew
That’s all I have time for this week.
Hi, Andrew,
First of all, thanks for the only free time you spent on
helping me, a poor knowledge guy in this area. I feel sorry for any
inconvenience that brought to your work and your deadline. You're selfless
and that makes me ashamed in terms of this. You can definitely ignore the
mail I sent and did your priority, but you didn't. I am not determined to
bother you and all the lammps users, developers. I* promise* these will be
the last questions in terms of this. It took 2 days before I determine to
send the inquire email, so please forgive me my ignorant and low-level
questions.
Then, your and lammps developers' guides are like the
lights before I finally get through the tunnel, though I can not still
fully understand all the suggestions you gave currently, I will study your
and lammps developers' suggestions line by line until I understand 100%.
You and lammps developers' suggestions will be always in my mind, for me
the suggestions not only help a lost Ph.D student to get confidence in
completing his program also establish the willingness to help other people
the way you did to me.
God bless you,
Hi, Andrew,
First of all, thanks for the only free time you spent on helping me, a poor knowledge guy in this area. I feel sorry for any inconvenience that brought to your work and your deadline. You’re selfless and that makes me ashamed in terms of this.
There is no need to be ashamed and there’s no need to apologize.
For someone in your situation, you are asking the right questions.
If I am too busy to provide more help, I will (usually) say so.
(But sometimes I am so swamped, I don’t even do that.)
Don’t feel bad for asking.
You have an obligation to your PI (if applicable) and your funding sources.
It’s just too bad there is nobody nearby who can help you.
Do the best you can.
You can definitely ignore the mail I sent and did your priority, but you didn’t. I am not determined to bother you and all the lammps users, developers. I promise these will be the last questions in terms of this. It took 2 days before I determine to send the inquire email, so please forgive me my ignorant and low-level questions.
Then, your and lammps developers’ guides are like the lights before I finally get through the tunnel, though I can not still fully understand all the suggestions you gave currently,
That is as it should be. My email contained a lot of information. You will have to click on the links to the LAMMPS documentation that describes what these fixes and commands do. Concepts (like thermostats, barostats, and the virial) are not specific to LAMMPS. They may require additional study by finding a good textbook on molecular dynamics. (The most popular textbooks that I know of are Frenkel&Smit and Allen&Tildesly, but perhaps there are newer books available now. But perhaps you don’t need to worry about buying textbooks now. Focus on your work.)
The most important suggestion was to start with a simple simulation that you understand. Then add complexity.
Cheers
Andrew
{kind=link}
{kind=link}