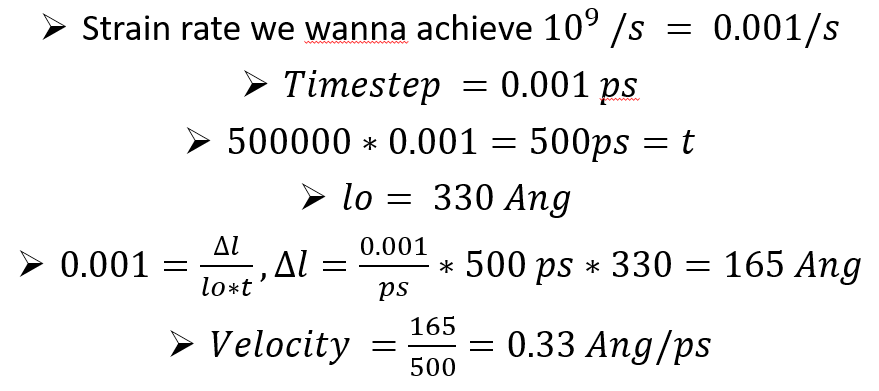Dear LAMMPS Users,
I use LAMMPS version: lammps- 7Aug, 2019. I downloaded it from the LAMMPS website. I am trying to achieve a strain rate of 0.001 /ps during the tensile test of the sss system described below. The length of the chosen system is approximately 330 Ang. The required velocity to achieve the aforementioned strain rate was calculated at 0.33 Ang/ps. The velocity calculations are shown below:
I am running the following code:
------------------------ INITIALIZATION ----------------------------
units metal
boundary s s s
atom_style atomic
----------------------- ATOM DEFINITION ----------------------------
lattice fcc 4.0896 origin 0 0 0 orient x -1 -17 8 orient y 1 -1 -2 orient z 7 1 3 spacing 18.8148877222268 2.44948974278318 7.681145747868608
region whole block 0 1 0 8 0 10.5 units lattice side in
create_box 1 whole
create_atoms 1 box
#------------------------ FORCE FIELDS ------------------------------
pair_style eam/alloy
pair_coeff * * Ag_Zhou04.eam.alloy.1 Ag
neighbor 1.5 bin
neigh_modify every 1 delay 1
reset_timestep 0
timestep 0.001
#------------------------- SETTINGS ---------------------------------
compute csym all centro/atom fcc
compute peratom all pe/atom
variable Lo_Fx equal 20
region Lo_Fx block INF INF INF INF INF ${Lo_Fx} units box
region Hi_Fx block INF INF INF INF 315 INF units box
region Fx union 2 Lo_Fx Hi_Fx
group Lo_Fx region Lo_Fx
group Hi_Fx region Hi_Fx
group Fx region Fx
group INT subtract all Fx
Store final cell length for strain calculations
variable tmp equal “lz”
variable L0 equal {tmp}
print "Initial Length, L0: {L0}"
variable tmpx equal “lx”
variable L0x equal {tmpx}
print "Initial Lengthx, L0x: {L0x}"
variable tmpy equal “ly”
variable L0y equal {tmpy}
print "Initial Length, L0y: {L0y}"
The velocity calculation that you showed is correct, even though there is a typo in the first line (0.001/s should be 0.001/ps).
Don’t you get total displacement Dl=165 Ang after 500000 steps? Your final lz should be 330+165 = 495 Ang.
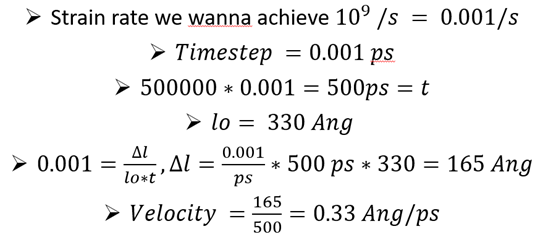
Dear Sir,
Unfortunately, no it goes much higher than expected. My Lz becomes ~575 Ang under 200000 steps for both the units lattice and box.
I am sorry about the typo, I didn’t realize that until your message. Any help in this regard would be appreciated.
Thank you
Best,
Ritesh
Did you try with group ‘INT’ in the ‘fix’ instead of ‘all’?
fix 1 all nvt temp 300 300 1
Sanjib
indeed fix nvt should only be applied to the INT group or else the computation of the temperature is flawed.
but that is not sufficient. you also need to run fix nve on group Hi_Fx and also zero the velocity of the Lo_Fx atoms.
it would be simpler, though, to skip the setforce and velocity command and simply have fix nvt on the INT group and then use fix move on the Hi_Fx group.
but also there is the question, why use shrinkwrap boundaries and a thermostat. with shrinkwrap boundaries you have an isolated object and that has not exchange of kinetic energy with a reservoir as a thermostat applies, so using fix nve would be more appropriate. or using periodic boundaries, w/o any fixed or pulled atoms and instead using fix npt (coupled only to x and y) and fix deform on z.
axel.
Dear Dr. Kohlmeyer and Dr. Chowdhury,
I appreciate both your help with this matter. It makes sense to apply nvt only on the INT region.
It is important to have shrink-wrapped boundaries because we are trying to achieve the activation of dislocation slip and twinning during the tensile testing. For the systems that we have periodic boundaries would prohibit the nucleation of trailing partial dislocation which is essential for the progression of dislocation slip.
However, I might have to revisit the application of temperature as you suggested, I think it might not be necessary. Additionally, I will restructure the code according to your suggestions.
I appreciate such a detailed explanation for my problem. Thank you once again.
Best,
Ritesh