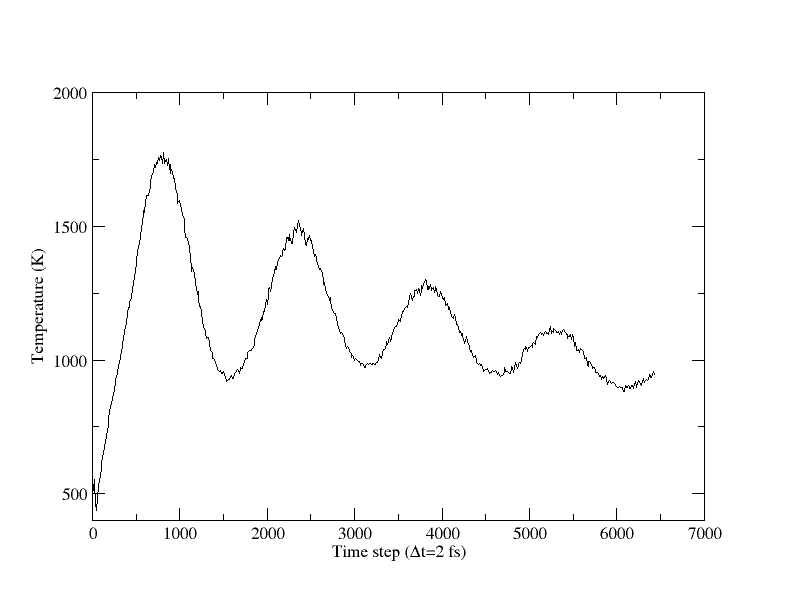
I attached the NPT input file to obtain the lattice constant of fcc-AlCu3 alloy using AlCu.eam.alloy, which is provided in Potential directory of LAMMPS. However, after initialization, the temperature is increased and the alloy is melted (crystal lattice is distroyed), which is viewed in xmovie. It's funny. In the case of pure Ni using Ni_us.eam, such problem is not occured.
Because I think the AlCu.eam.alloy potential is reliable, maybe are there any further command options for alloy simulation?
I would make sure you are getting the crystal structure you
expect with the Al and Cu in the right places. Using lattice
fcc in the way you are w/out knowing where LAMMPS is putting
the basis atoms, might be problematic.
In my understanding, in fcc crystal lattice (four basis atoms), first basis atom is located at position (0 0 0) -Al and others are located at (0.5 0.5 0.0), (0.5 0.0 0.5) and (0.0 0.5 0.5) -Cu3 in the unit cell.
If my setting way to position the basis atoms is not right, how can I do correctly in LAMMPS?
That looks right - but you'd have to look inside the code to
figure that out. So normally, you'd define those
basis atoms yourself in a lattice custom command rather
than assume an ordering used internally by keyword fcc.
The problem with your simulation is that the temperature
shoots up so high and then isn't relaxing to 500K. So
you're probably melting the crystal. Your relaxtion
time for temperature is quite long - 10.0 = 10 psec =
5000 timesteps, so I'd try playing with that and not
use the drag keyword initially.
Another thought - can you run NVT of your initial system and get a stable
temperature? If so, then your NPT run must be changing the box volume
dramatically which indicates a lattice constant that doesn't match what
the potential wants to induce.
The system evolves towards 500k. However the system is not crystal but the liquid. That is, the liquid after some initial time step goes to the 500K. Even if I do not use the drag, the system becomes the liquid soon.
The effect of NVT is same to NPT. When I tested the smaller lattice constant, then the system was expanded quikly and then became liquid again. Moreover this potential was developed for fcc crystal and its lattice constant is 4.5A.
When I tested the pure metal such as fcc-Ni using the Ni-u3.eam, I did not meet such problem and got the equilibrium lattice constant agreed with the experimental value. Whenever I tried to treat the alloy system such as AlNi and AlCu, I had to meet such trouble.
You r script uses a lattice constant of 4.2, not 4.5. If you're building
the lattice you think you should (spacing, structure) which you probably
need to viz to make sure, and you can't run NVT and keep it stable
at a T well below the melting point, then I'd say the potential is not
formulated for the system you think it is. Are you sure the AlCu
potential is meant for AlCu3 ? Are there other AlCu alloys?
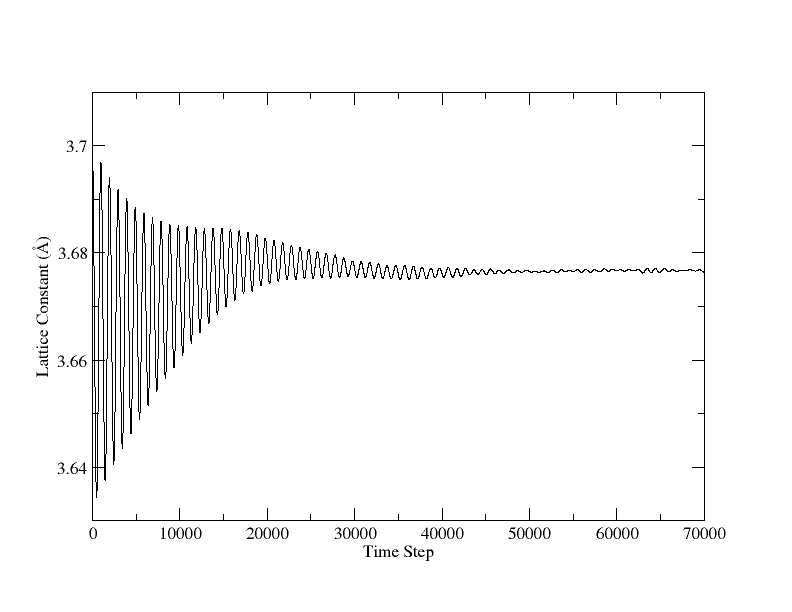
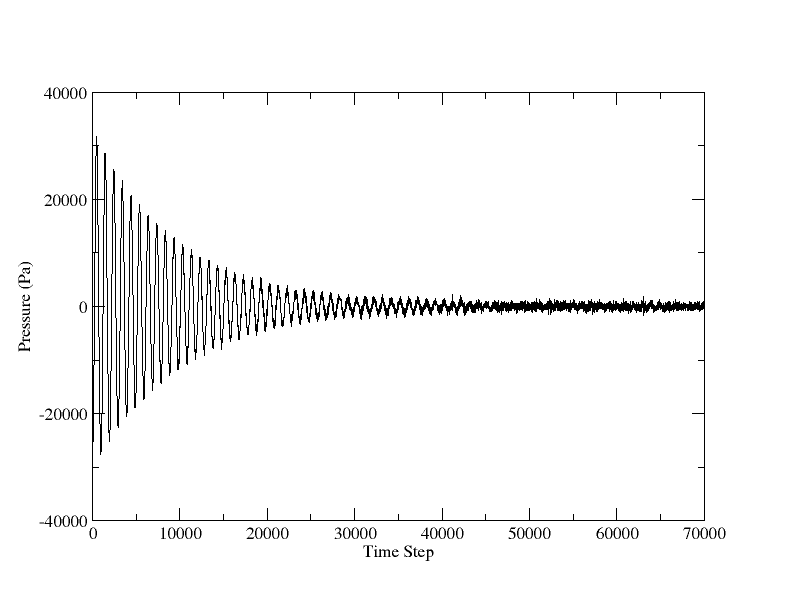
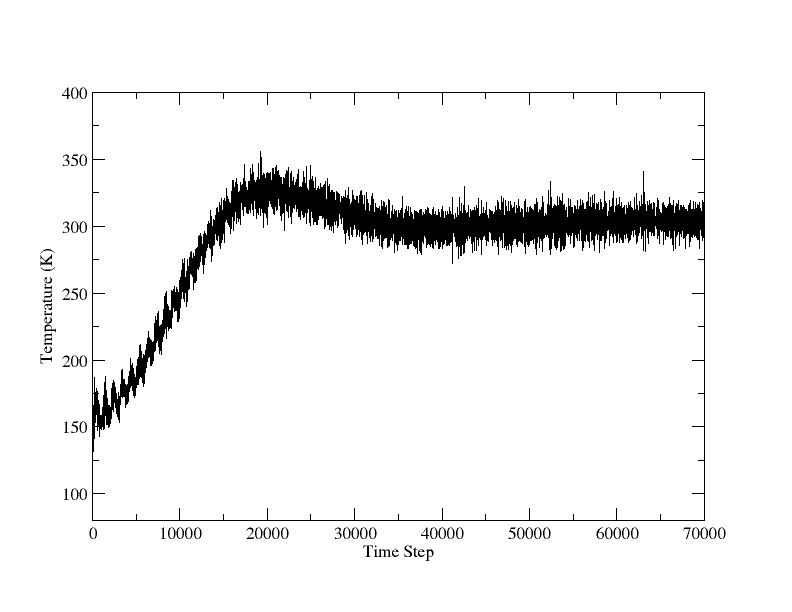
thank you very much for your many comments and hints. After viewing the paper for AlCu3, I found the lattice constant of fcc-AlCu3 is 3.71 A. Then the test of NPT of AlCu3 reached reasonable results, attached.
So it seems that MD simulations would sensitive to the lattice constant. This means, if the bad lattice constant is used to determine the true lattice constant, the simulation could be failed? How much deviation from the true lattice constant can we start?
So it seems that MD simulations would sensitive to the lattice constant.
This means, if the bad lattice constant is used to determine the true
lattice constant, the simulation could be failed? How much deviation
from the true lattice constant can we start?
Solids are very sensitive to the lattice constant. A percent or two is
a big effect. Your simulation is off by 15% o rmore, so the potential
is way off. Look at thermal expansion coefficients to see how little
a solid expands across a wide temperature range.