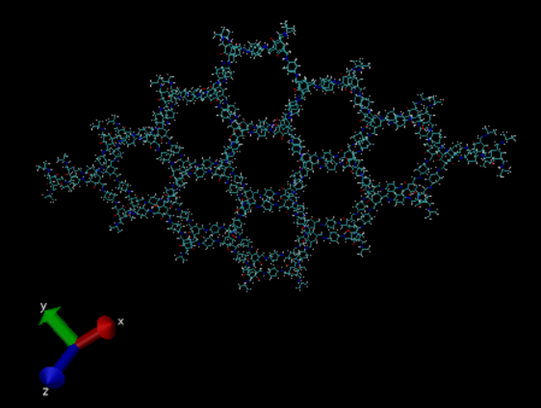
Hello,
I am simulating a bilayer TpPa-1 COF system (please see pic1) to check the gap between the layers at equilibration. I have simulated the system with “lj/class2” and “reactive” force fields separately. Using lj/class2, the system completely collapsed after the 5ps equilibrium simulation (please see pic2). With reax ff, the bilayer system somehow keeps the COF structure after the 20 ps simulation (please see pic3).
My question is why the bilayer system quickly collapses with lj/class2 setting. May I have your advice on this?
The input script is given below:
units real
dimension 3
boundary p p p
atom_style full
read_data Myfile
group carbon type 1
group oxygen type 4
group hydrogen type 2
group nitrogen type 3
pair_style lj/class2/coul/long 10.0
pair_coeff 1 1 0.10458 3.431 #lj/cut/coul/long
pair_coeff 2 2 0.04390 2.571
pair_coeff 3 3 0.0689 3.261
pair_coeff 4 4 0.05958 3.110
pair_modify mix arithmetic
bond_style class2
bond_coeff 16 1.0 100.0 80.0 80.0
angle_style class2
angle_coeff 115 109.5665 105.3402 0 0
angle_coeff 115 bb 10.5872 1.0119 1.5228
angle_coeff 115 ba 3.6551 24.895 1.0119 1.5228
fix tmp_rescle all temp/rescale 1 300.0 300.0 1.02 1.0
#velocity all create 300.0 34387 rot yes dist gaussian # for water
kspace_style ewald 1.0e-6
minimize 1.0e-4 1.0e-6 100 1000
*************** Setting ******************************
timestep 0.1
fix fxlan all langevin 298.0 298.0 1000.0 48279
fix fxnve all nve # ← needed by fix langevin (see lammps documentation)
thermo 100
run 2000
unfix fxlan
unfix fxnve
neighbor 4.0 bin
neigh_modify delay 0 every 10 check yes page 500000 one 50000
reset_timestep 0
compute teatom all temp
compute peatom all pe/atom
compute keatom all ke/atom
thermo 500
thermo_style custom step temp pe etotal press
thermo_modify norm no flush yes
fix nvt_ensemble all nvt temp 300.0 300.0 1000.0
variable end equal 50000
dump pos all custom ${end} pos_filename id element type q x y z
dump_modify pos format line “%5d %5s %d %13.10f %17.12f %17.12f %17.12f”
dump_modify pos sort id
dump_modify pos element C H N O #C C C C C C O H N
dump vel all custom ${end} vel_filename id element vx vy vz
dump_modify vel format line “%5d %5s %18.15f %18.15f %18.15f”
dump_modify vel sort id
dump_modify vel element C H N O #HW OW Cl Na
dump trj1 all atom 100 wat.trj
dump trj all custom 500 wat-.dat id element type x y z vx vy vz fx fy fz
dump_modify trj sort id
dump_modify trj element C H N O #HW OW Cl Na
#write_data waterinfo.data
write_restart waterinfo.restart
timestep 1.0
run 50000
pic1
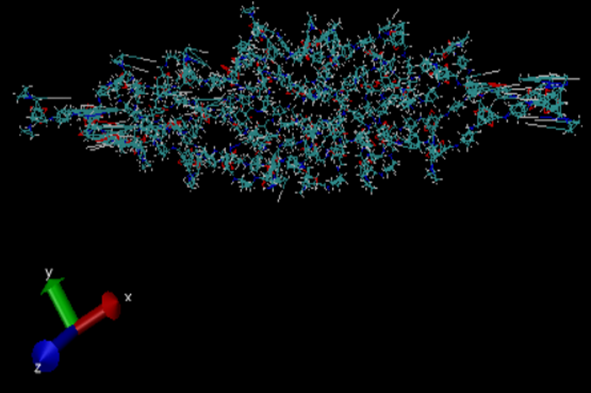
pic2
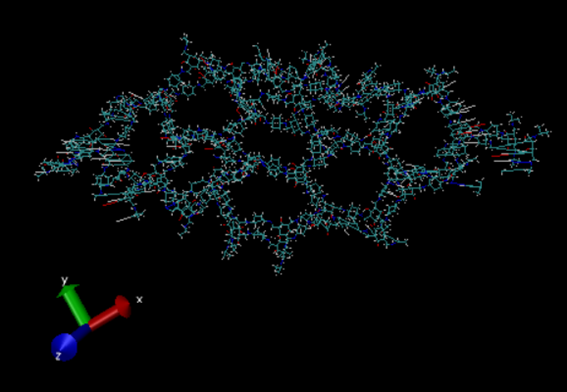
pic3