Hello. I am trying to model a carbon nanotube unit cell (see attachment please). Most importantly, we want the tube is continuous through boundaries. That is, the top layer of carbon atoms should be bonded with the bottom atoms in a neighboring unit cell. But problem is: how to model those bond in a data file.
For example, in the attached figure, atom1 should be bonded with atom2 in the upper unit cell. In the input file, however, I cannot directly bond atom1 and atom2 because the system doesn’t know atom2 is from the present unit cell or from the upper unit cell. So how can I do it. Thanks.
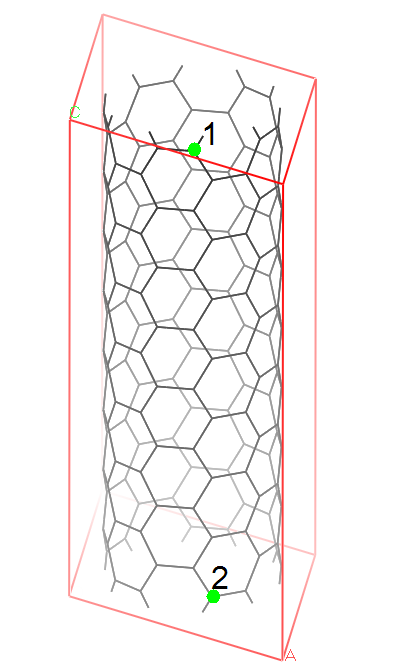
Hello. I am trying to model a carbon nanotube unit cell (see attachment
please). Most importantly, we want the tube is continuous through
boundaries. That is, the top layer of carbon atoms should be bonded with the
bottom atoms in a neighboring unit cell. But problem is: how to model those
bond in a data file.
just normally. if you are using periodic boundary conditions
and the correct box size, atom 1 and atom 2 are actually next
to each other!
cheers,
axel.
Thanks Axel. Yes, Periodical Boundary can make the tube looks continuous at the initial state. But, if there is no bonding across PB, after a while of dynamic simulation, the tube could lose the continuity (imagine somehow the tube is rotated about the z axis, then how could the continuity be hold?), especially when the temperature is high or there is loading.
So I guess my question was not clear. What we want is not only continuity in the initial state, but also more importantly, the connectivity between the two ends to ensure all the time continuity.
Can LAMMPS do what we want to do? More specifically, we want to include the bonding between the two ends in the data file - bonds section, but direct input of atom1-atom2 will confuse the code because atom1 is actually bonded to atom2 in the next unit cell. Please give me a hint. Thank you very much. 
Ling
Have you actually tried putting those bonds in the data file? I have had no problems with LAMMPS being confused about which image of the atoms to use when the bonds were correctly defined and the atoms were adjacent in a minimum image calculation. The simulation doesn’t “know” about boxes because minimum image takes care of that problem. After the first few steps, atom1 and atom2 may be in the same box, so why would being the same distance apart across an imaginary line matter? It doesn’t and if you try putting those bonds in the data file, you will see that while you may be confused, LAMMPS will not be.
Joanne
If you have a linear molecule of 5 atoms 1-2-3-4-5
in a periodic box such that 1 and 5 should be bonded
across the periodic boundary, then just put 5 bonds
in your data file: 1-2, 2-3, 3-4, 4-5, 1-5. LAMMPS will
do the right thing.
Steve
Thank you all for your help.
You are right. LAMMPS will not be confused by what confused me. A trial job has confirmed the effectiveness of the minimum image calculation in LAMMPS. LAMMPS is proved to be smarter than me. Appreciate your replies and generous help.
Ling