Hello LAMMPS users, I am using windows 30 july 2021. units are real. I have a query regarding rigid command.
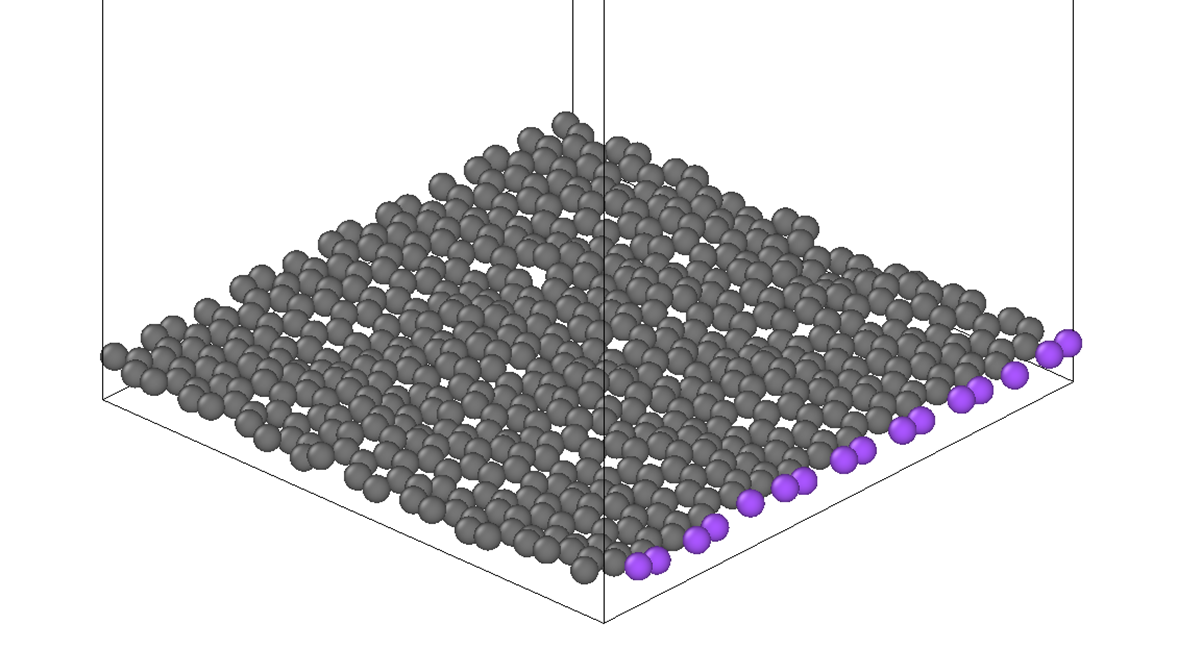
I want to move graphene as a rigid body. For this I have created two groups. One group is fixedatoms (purple colored) and the second group is rigidcarbonatoms (grey color).
For this, purple colored atoms are displaced by some displacement by using command.
“displace_atoms fixedatoms move 0.001 0 0 units box”
And as i want to do this at fix temperature. I am using command for rest atoms as given below
“fix 3 rigidcarbonatoms rigid/nvt single temp 300 300 100”
But here there is stretching between bonds between purpled and grey carbon atoms (adjacent to purpled color atoms).
Is there any way that this stretching does not happen, if i move purple colored carbon atoms?
Should I use this command as;
“fix 1 fixedatoms rigid single”
“displace_atoms fixedatoms move 0.001 0 0 units box”
“fix 3 rigidcarbonatoms rigid/nvt single temp 300 300 100”
But here still how to overcome stretching that dont take place when I move purple colored atoms.
Thank you,
Ankit
There are three different issues here:
- when you define rigid bodies, it will be the fix that maintains information about position and orientation of the rigid body. thus if you are then modifying positions of individual constituent atoms in the rigid body, you will have to rebuild/reinitialize the affected rigid bodies, which will not happen by default. please see the documentation for details.
- you cannot have a (single) rigid object and have it at the specific temperature at the same time. the temperature that you control is the kinetic energy of the rigid body itself (translation and rotation), but for a single object that is ill defined.
- it is impossible to comment on the “bonds” without knowing more about what information you write out and how you visualize it. most likely you are not providing any bond information to the viz tool and it will just generate them from a simple distance heuristic and may thus be completely bogus.;
axel.
There’s a command that might be suitable for you that results in that entire group is feeling the same force, i.e. you should explore that
https://docs.lammps.org/fix_aveforce.html
Thank you, Lukasz. I will look into it.