Hello everybody,
because I faced some problems with respect to the EAM-MD simulation for alloy, e.g. Ni-Al, I would like to post this email to LAMMPS community in order to get any advices and comments or hints.
1) Solid:
When I tried to simulate the fcc crystal at several temperatures, the undesirable melting is occured in the case of Ni3Al fcc crystal, that is, it melts at even 900 K which is much lower than the experimental value, 1663 K. Meanwhile in the case of NiAl3 fcc crystal, I obtained the reasonable melting temperature (1100 K, exp 1120K) using the so-called "void" method (Chem. Phys. 224[1997], 253-261). I am using Y. Mishin potential (Acta Mater. 52[2004], 1451-1467). Where is such deviation from?
2) Phase separation in liquid:
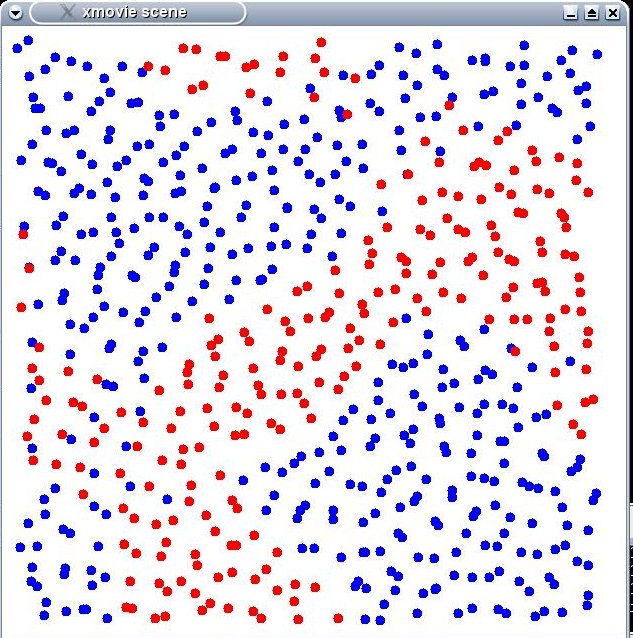
To get the atomic configuration of liquid Ni25Al75, I performed the NPT simulation starting from fcc crystal and continue the NVT simulation to equilibrate the system. At the end of NPT or NVT simulation, I got also undesirable configuration, that is, phase-separated liquid, not fully randomized mixture of phase. Dump file shows visually that there exist some separated part of pure Ni and pure Al. Is it impossible to get fully mixed configuration of alloy liquid by NPT or NVT MD simulation? If not so, how can we get liquid configuration in fully mixed state? Or is such problem from the EAM potential used in work?
3) Not conserved thermodynamic quantities in NVE simulation for liquid:
When I used the not mixed but phase separated configuration of alloy liquid as initial configuration for NVE simulation, the total energy was not conserved but increased, and accordingly the temperature was also increased. The pressure is also increased. However, when I follow the same procedure for pure Ni (Acta Mater 47[1999], 3181-3187), the quantities were conserved well in NVE simulation of liquid. Where is such deviation from? Is it due to the bad initial configuration (separated configuration)?
Any advices and comments or hints would be thankful!
Best,
Chol-Jun