First off, you need to read the manual even more carefully, and you probably need somebody local with more experience in programming to help you get a better understanding of how to write and debug programs. writing this kind of complex input is essentially writing a little program in the “LAMMPS input language” and thus you need to apply concepts of programming and debugging to get meaningful results and have an effective approach to implementing what you need. these are skills that are next to impossible to teach via email.
there is a technical issue (your equal style expression use the wrong kind of variable reference and thus it will always give the same result) and a conceptual problem. please note that equal style variables in LAMMPS are more like functions than like variables.
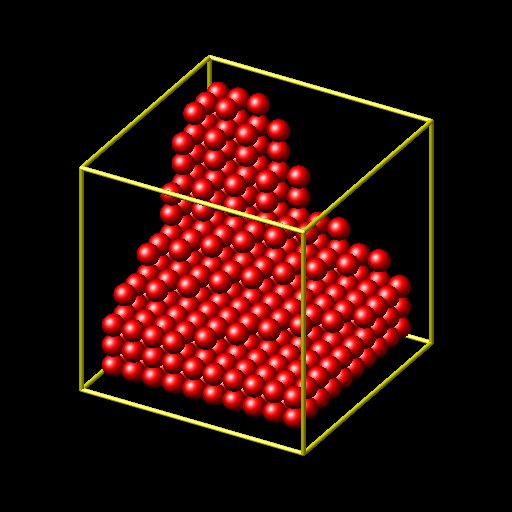
the “var” option to the create_atoms command can be used to select on which lattice positions atoms will be created. this means the “variable” pointed to by the “var” keyword is evaluated for each potential set of coordinates within a region (as determined by the lattice command and the region command) and when it value is non-zero, the atom will be created.
thus the “internal” variables are not an input that needs to be set but they will be set internally (hence the name) by LAMMPS and then the equal style variable is consulted on whether those positions would return a zero or non-zero value. the following input
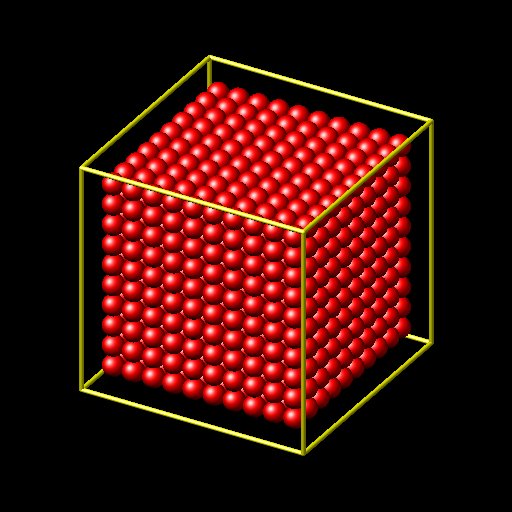
region cube block 0 10 0 10 0 10
create_atoms 1 region cube
will create a cube filled with atoms. however when using this input with the var keyword
variable xx internal 0.0
variable yy internal 0.0
variable zz internal 0.0
variable select equal sqrt(v_xxv_zz+v_yyv_zz)<5
region cube block 0 10 0 10 0 10
create_atoms 1 region cube var select set x xx set y yy set z zz
it will create a partially filled cube with the function in the equal style variable “select” determining which of the possible lattice positions will be created.
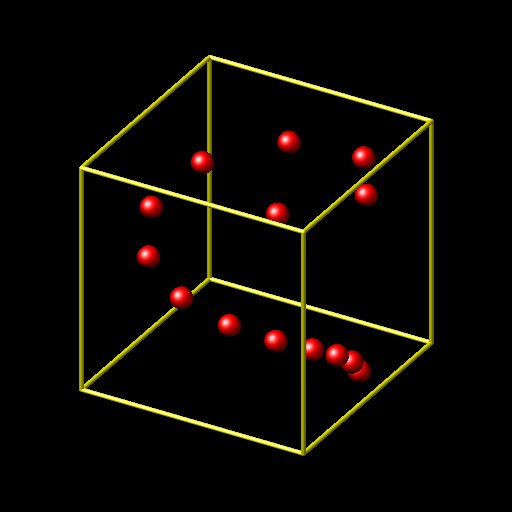
what your input is doing is creating multiple atoms at exactly the same location. that is definitely not what you want.
if you want to create atoms in a spiral on a cylindrical surface, you have to follow a very different approach.
a) you should be using create_atoms single
b) you should be using 3 equal style variables to compute the x, y, and z position from within a loop based on the loop index.
here is an example:
variable i loop 14
variable radius index 5.0
variable rotate index 3.0
variable transl index 0.7
variable z equal v_iv_transl
variable x equal v_radiussin(v_rotatev_iPIv_z/180.0)+5.0
variable y equal v_radiuscos(v_rotatev_iPI*v_z/180.0)+5.0
label loop
create_atoms 1 single {x} {y} ${z}
next i
jump SELF loop
however, if you want to use those as tether positions for your chains, then you have much more work to do. and that goes beyond the scope of this mailing list exceeds the amount of time I can spend to solve your problem for you. so please see my advice from the beginning.
axel.

{kind=link}