Hello everyone,
I use compute cna/atom to identify FCC and surface atoms in a Nickel single crystal for a crack propagation problem. I have the following problems while running the simulation.
- Except for the first dump, the compute cna/atom output is random/garbage for subsequent timesteps on multiple processors. (I saw that this problem has been reported earlier)
- Even on one processor, the atoms that are correctly identified as “fcc” and “unknown” in the first step, have the same tag till the end of the simulation even though more atoms should become “unknown” as the crack advances.
There was a similar problem with the compute ackland/atom command too.
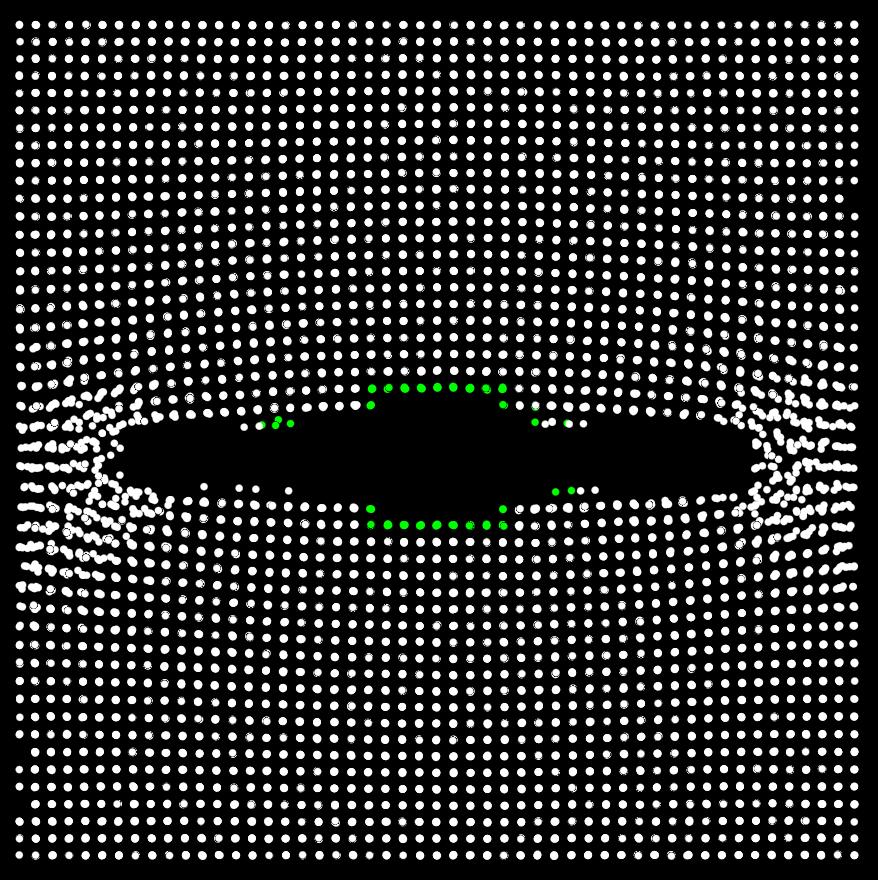
The first two pictures are snapshots of a simulation on a single processor that show the atoms before appying a deformation (cna_ti.jpg) and another after the crack has advanced substantially (cna_tf.jpg). Green atoms refer to “unknown” atoms and white atoms refer to “fcc” atoms. The atoms that are green initially, stay green throghout. It is as if the cna output for an atom does not get updated.
I have tried changing the cna cutoff slightly as well as trying out different frequencies of building the neighbor lists.
Thanks,
Santosh