Sorry Axel, I maybe wasn’t precise enough in my original email. Really, the main issue I wanted to bring up is that while both fix qeq/comb and fix qeq/dynamic reference the Rick, Stuart, Berne paper, they don’t actually follow the methods outlined in that paper. I don’t think the documentation really explains what those fixes are doing. I suppose my reason for writing the email was to say I think the documentation should be more explicit about what is being done in those fixes, and to understand why they suggest that extended Lagrangian methods are implemented; none of the fixes on the fix qeq/point doc-page are extended Lagrangian methods, they are all some form of minimization.
In regards to the COMB energy drain, I didn’t mention it as a request for help trouble shooting. Based on previous mailing list discussions (https://sourceforge.net/p/lammps/mailman/message/36666819/) and discussions with a member of Susan Sinnott’s group this seems to just be a fact of COMB. I only mention it because I don’t know that I’ve ever been fully satisfied with the response I’ve seen that suggests the loss in energy is solely due to the charge dynamics, which as implemented are no more coupled to the nuclear dynamics than the matrix inversion method and therefore shouldn’t cause extremely rapid energy drain unless the settings are poorly chosen as you mention. But the responses that I’ve received from the Sinnott group is that COMB does have energy drain, and you need to use a thermostat to reach a steady state where the thermostat feeds energy in fast enough to counterbalance the energy drain.
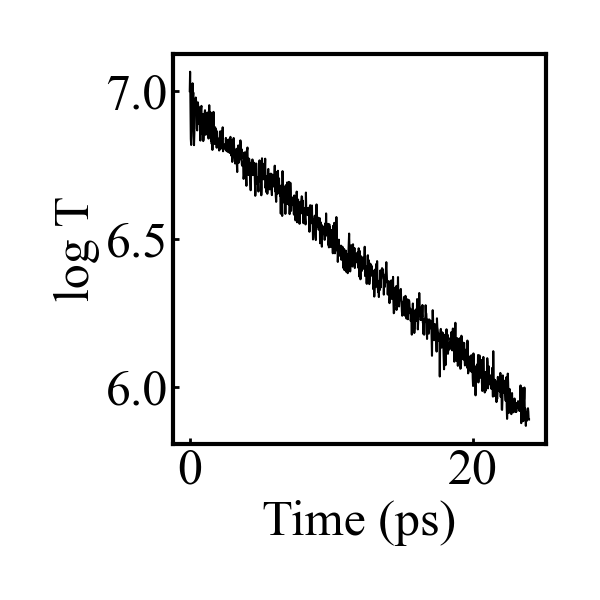
I’m finishing up my dissertation over the next couple months, but after I get settled in my new job in the fall I intend to take a deeper dive into the energy drain and related issues I’ve seen with COMB. Hopefully I can give a fuller picture then. As a quick example, though, here are the results for the bulk corundum simulation given below:
Even with a timestep of 0.05 fs, equilibrating the charges every timestep, and a tolerance of 0.00001 for the charge equilibration, there is an exponential decay in the Temperature (and total energy) once the thermostat is removed. Maybe I missed something in my simulation setup, but I don’t think there is anything in my simulation that would lead to this kind of exponential energy drain.
The figure is the log of the temperature after the thermostat is removed.
#simulation of corundum using COMB3
units metal
atom_style charge
dimension 3
boundary p p p
read_data data.small_corundum
variable comx equal vcm(all,x)
variable comy equal vcm(all,y)
variable comz equal vcm(all,z)
pair_style comb3 polar_off
pair_coeff * * ffield.comb3 O Al
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
timestep 0.00005
thermo_style custom time temp press pe ke etotal v_comx v_comy v_comz
thermo 500
thermo_modify norm yes
fix 0 all qeq/comb 10 0.01
velocity all create 1043.0 42128 dist gaussian rot yes mom yes
minimize 1.0e-3 1.0e-3 100 1000
unfix 0
neigh_modify every 10 delay 0 check yes
fix 1 all qeq/comb 1 0.00001
fix 2 all nve
fix 3 all temp/berendsen 1043 1043 0.1
run 10000
reset_timestep 0
dump coords all custom 500 al_coord.lammpstrj id type q x y z
dump_modify coords sort id
unfix 3
velocity all zero linear
velocity all zero angular
run 500000