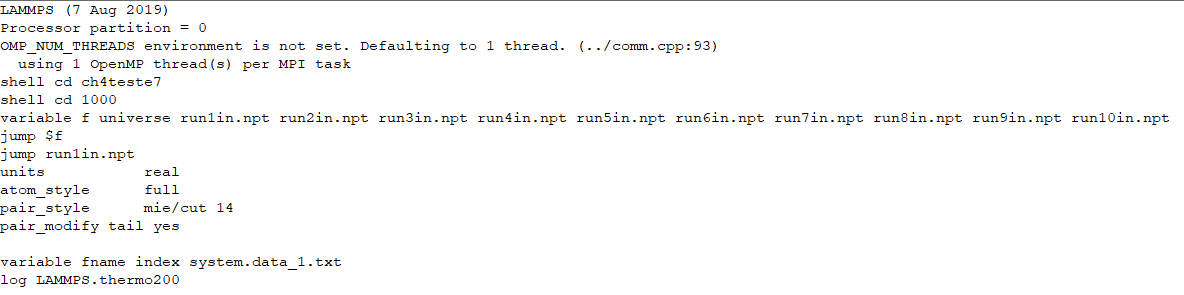
Dear colleagues,
I have questions about the LAMMPS -partition flag and “how to multiple”. I am using version 7Aug19.
Suppose I have 10 complete scripts (run1in.npt … run10in.npt) in a given directory.
I would like to run 2 simulations at a time in this universe of scripts, and after each one is finished, continue running the others.
To run 2 simulations at a time I use the “-partition 2xN” flag, where N is the number of processors in the partition.
My idea is to create an intermediate script (inputs.npt), with variables of type universe (as suggested in the manual (https://docs.lammps.org/Howto_multiple.html)
I’m making mistakes because I didn’t quite understand the concept of “how to multiple”.
When I run the intermediate script (inputs.npt) with the commands below:
variable i universe run1in.npt run2in.npt run3in.npt run4in.npt run5in.npt run6in.npt run7in.npt run8in.npt run9in.npt run10in.npt
jump $i
And the execution:
mpirun -np 2N ./lmp_mpi -partition 2xN -in inputs.npt
LAMMPS only runs normally the run1in.npt and run2in.npt files, not going on to the others (run3in.npt run4in.npt… run10in.npt).
Could someone help me to indicate what the probable mistake I made and a possible solution to run the 10 scripts in a sequence of 2 in 2 simulations?
Thanks in advance for your time.
Best Regards,
Emerson Parazzi Lyra
PhD candidate
School of Chemical Engineering
University of Campinas
500 Albert Einstein Ave, Campinas, SP, 13083-852, Brazil
Email: [email protected]
Orcid ID: https://orcid.org/0000-0002-7969-3764