Dear LAMMPS users,
I’m using LAMMPS (3 Mar 2020) with rigid, dipole, molecule and misc package. I have compiled LAMMPS with serial by now, not with mpi (eventually I would like to do it for mpi)
-
Which flags do I have to use to find the full neighbor list when I use PBC conditions? As you can see in the code below I already use the request for the full list. I’m using in the LAMMPS script neighbor 0.0 bin and no other neighbors modify by now
-
Also what should be the proper way to set the neighbor list cutoff? Would the command neighbor bin be the correct one for this purpose?
If you need LAMMPS script, Initial Snapshot or anything else, please let me know
I send you more details in case that you need it:
I’m trying to implement active matter but with local momentum conservation with a specific rule. Imagine that I have a system with particles of type i (input, let say 1) and one particle of type j (input, let say 2. This is actually a rigid spherical colloid made of particles that have one angle bond with the type 2 particle in the center and two particles in the edge of the colloid to define an axis). I want to apply a force F_a (input, around 300 in lj units) in the particle of type 2 (in the direction of the axis defined by the angle bond) and apply a force F_p (computed in the program with a specific rule) on each neighbor (type 1) of this particle such
sum over all neighbors of type 1 (F_p) = - F_a
This way sum(F)=F_a-F_a =0 all time (except numerical fluctuations) and local momentum will be conserved with this redistribution of the force. I find a similar post in the mail list but it doesn’t solve my problem
https://lammps.sandia.gov/threads/msg26076.html
I already implemented a code to do that but use my own list of neighbors “by hand” (with two loops over all atoms, computing the distance and with a cutoff… not optimal…) and works properly. The problem is that I have tried to do the same using LAMMPS’ neighbor list and something happens when the list has to use PBC, but with no PBC the code works. When the particle of type 2 is close to the boundary (this is, when the cutoff for the list is more than the distance of the particle to the boundary) the sum over all neighbors (F_p) != - F_a as you can see in the figure below (I show the modulus of the force here)
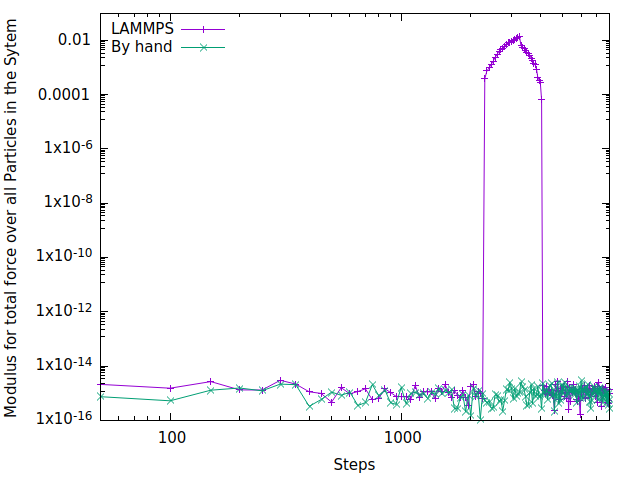
I have compared the codes for the same cutoff (3.0 by hand and I suppose that 3.0 if lammps takes the neighbor list cutoff from the pair_style + skin distance) and it looks like the neighbors in the boundary are not exactly the same, and that is why the force is not zero, that’s mean that I’m doing something wrong with the PBC conditions…
Just to be sure I also set
neigh_modify every 1 delay 0 check no
and the neighbor list given by lammps had more neighbors but still not all of which were present in the “by hand” neighbor list
Thank you very much
ciao
José
/////////////////////////////////////////////////////////////////////////////////
//////////////////// fix_code.cpp //////////////////////////////////////
/////////////////////////////////////////////////////////////////////////////////
#include “domain.h”
#include “math.h”
#include “stdlib.h”
#include “string.h”
#include “fix_puller.h”
#include “atom.h”
#include “force.h”
#include “neighbor.h”
#include “neigh_list.h”
#include “neigh_request.h”
#include “group.h”
#include “update.h”
#include “error.h”
using namespace LAMMPS_NS;
using namespace FixConst;
/* ---------------------------------------------------------------------- */
FixPuller::FixPuller(LAMMPS *lmp, int narg, char **arg) : Fix(lmp, narg, arg) {
if (narg == 9){
type1 = force->numeric(FLERR, arg[3]);
type2 = force->numeric(FLERR, arg[4]);
type_angle = force->inumeric(FLERR, arg[5]);
cutoff = force->numeric(FLERR, arg[6]);
Pe = force->numeric(FLERR, arg[7]);
Rc = force->inumeric(FLERR, arg[8]);
A=1;
B=0;
alpha=1;
beta=1;
}
else if (narg == 13){
type1 = force->numeric(FLERR, arg[3]);
type2 = force->numeric(FLERR, arg[4]);
type_angle = force->inumeric(FLERR, arg[5]);
cutoff = force->numeric(FLERR, arg[6]);
Pe = force->numeric(FLERR, arg[7]);
Rc = force->inumeric(FLERR, arg[8]);
A = force->inumeric(FLERR, arg[9]);
B = force->inumeric(FLERR, arg[10]);
alpha = force->inumeric(FLERR, arg[11]);
beta = force->inumeric(FLERR, arg[12]);
}
else{
error->all(FLERR, “Illegal fix hy/puller command”);
}
force_reneighbor = 1;
next_reneighbor = -1;
}
/* ---------------------------------------------------------------------- */
int FixPuller::setmask(){
int mask = 0;
mask |= POST_FORCE;
return mask;
}
/* ---------------------------------------------------------------------- */
void FixPuller::init_list(int /id/, NeighList *ptr)
{
list = ptr;
}
/* ---------------------------------------------------------------------- */
void FixPuller::init() {
int irequest = neighbor->request(this,instance_me);
neighbor->requests[irequest]->pair = 0;
neighbor->requests[irequest]->fix = 1;
neighbor->requests[irequest]->half = 0;
neighbor->requests[irequest]->full = 1;
}
/* ---------------------------------------------------------------------- */
void FixPuller::setup(int vflag)
{
if (strstr(update->integrate_style,“verlet”))
post_force(vflag);
}
/* ---------------------------------------------------------------------- */
void FixPuller::post_force(int vflag){
////////////// For the PBC in the colloid axis
double *boxlo = domain->boxlo;
double *boxhi = domain->boxhi;
double x_size = boxhi[0] - boxlo[0];
double y_size = boxhi[1] - boxlo[1];
double z_size = boxhi[2] - boxlo[2];
/////////////////////////////////////////////
int i,j,ii,jj,inum,jnum,itype,jtype;
double xtmp, ytmp, ztmp;
int *ilist, *jlist,*numneigh, **firstneigh;
int i1,i2,i3,n;
double delx,dely,delz;
double rsq,r;
double **x = atom->x;
double **f = atom->f;
double **mu = atom->mu; // Used as auxiliar variable
int *type = atom->type;
int atype;
int **anglelist = neighbor->anglelist;
int nanglelist = neighbor->nanglelist;
double Fa;
double Fa_x, Fa_y, Fa_z;
int nlocal = atom->nlocal;
int newton_bond = force->newton_bond;
int max_num_neigh=20000;
int list_neigh[max_num_neigh], counter; //Auxiliar list
double norma, weight_factor[max_num_neigh], prod_scal, cos_theta_2;
for (n = 0; n < nanglelist; n++) { //[FOR in nanglelist]
atype = anglelist[n][3];
if(atype == type_angle){ // [IF angle types]
i1 = anglelist[n][0];
i2 = anglelist[n][1];
i3 = anglelist[n][2];
delx = x[i3][0] - x[i1][0];
if (delx > x_size * 0.5) delx = delx - x_size;
if (delx <= -x_size * 0.5) delx = delx + x_size;
dely = x[i3][1] - x[i1][1];
if (dely > y_size * 0.5) dely = dely - y_size;
if (dely <= -y_size * 0.5) dely = dely + y_size;
delz = x[i3][2] - x[i1][2];
if (delz > z_size * 0.5) delz = delz - z_size;
if (delz <= -z_size * 0.5) delz = delz + z_size;
rsq = delxdelx + delydely + delz*delz;
r = sqrt(rsq);
Fa = Pe/1.;
Fa_x = Fadelx/r;
Fa_y = Fadely/r;
Fa_z = Fa*delz/r;
if (newton_bond || i2 < nlocal) {/// [IF bonds]
f[i2][0] += Fa_x;
f[i2][1] += Fa_y;
f[i2][2] += Fa_z;
mu[i2][0] = Fa_x;
mu[i2][1] = Fa_y;
mu[i2][2] = Fa_z;
}/// [IF bonds]
}/// [IF angle types]
} /// [FOR in nanglelist]
inum = list->inum; // # of I atoms neighbors are stored for
ilist = list->ilist; // local indices of I atoms
numneigh = list->numneigh; // # of J neighbors for each I atom
firstneigh = list->firstneigh;// ptr to 1st J int value of each I atom
for (ii = 0; ii < inum; ii++){ // [FOR ii]
i = ilist[ii];
itype = type[i];
if(itype == type1){ //[If typei and type1]
xtmp = x[i][0];
ytmp = x[i][1];
ztmp = x[i][2];
jlist = firstneigh[i];
jnum = numneigh[i];
norma = 0;
counter = 0;
for (jj = 0; jj < jnum; jj++) { // [FOR jj]
j = jlist[jj];
j &= NEIGHMASK;
jtype = type[j];
if (jtype == type2){ // [If typej and type2]
delx = x[j][0]-xtmp;
dely = x[j][1]-ytmp;
delz = x[j][2]-ztmp;
Fa_x=mu[i][0];
Fa_y=mu[i][1];
Fa_z=mu[i][2];
rsq = delxdelx + delydely + delz*delz;
r=sqrt(rsq);
list_neigh[counter]=j;
/// Redistribution Rule
prod_scal = delxFa_x + delyFa_y + delzFa_z;
cos_theta_2 = sqrt(0.5prod_scal/(r*Fa) + 0.5);
weight_factor[counter] = (cutoff - r)*cos_theta_2;
norma += weight_factor[counter];
counter++;
} // [If typej and type2]
} // [FOR jj]
for (jj = 0; jj < counter; jj++) { // [Redistribution Loop]
j=list_neigh[jj];
f[j][0] -= Fa_xweight_factor[jj]/norma;
f[j][1] -= Fa_yweight_factor[jj]/norma;
f[j][2] -= Fa_z*weight_factor[jj]/norma;
} // [Redistribution Loop]
} //[If typei and type1]
}// [FOR ii]
}