Dear colleagues,
I am getting the energy from the van der Waals potential of a specific configuration of a methane pair through the “run 0” command.
My idea is to compare the energy value of this configuration obtained in Lammps with the energy provided by ab initio calculations (in order to obtain lennard jones parameters for a force field).
However, I believe that I made a mistake in my energy calculations (van der Waals), because when testing values of the OPLS force field parameters, I have found discrepancies between the energy obtained in the Lammps and in my external code (to calculate Lennard-Jones potential energy). The error is obviously in my external code or in my Lammps script to calculate the energy (I’m trying to figure out). I’m currently trying to understand how Lammps calculated the energy (van der Waals) of the methane pair.
I know from the special_bonds command (https://lammps.sandia.gov/doc/special_bonds.html) that, by default, all 3 Lennard-Jones and 3 Coulombic weighting coefficients = 0.0. So I would have 25 non-bonded interactions for 2 pairs of methane (i.e., C1C2; C1H5; C1H6; C1H7; C1H8; C2H1; C2H2; C2H3; C2H4; H1H5; H1H6; H1H7; H1H8; H2H5; H2H6; H2H7; H2H8; H3H5; H3H6; H3H7; H3H8; H4H5; H4H6; H4H7; H4H8).
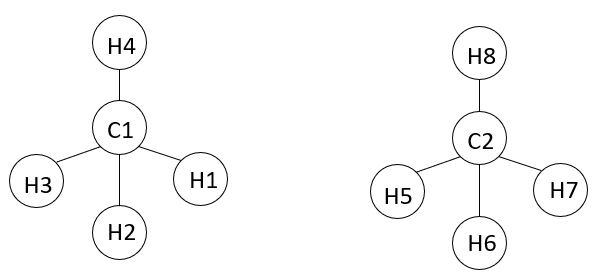
Attached I send the file (an example) to obtain the energy of 1 pair of methane in Lammps (runvdwl13in.npt) and an auxiliary topology file which I read to run the Lammps script (system13.data.txt). I also send the attachment of the log file obtained by Lammps (logvdwl.system.data.txt_13). I used the command “thermo_style custom evdwl” to calculate van der Waals energy for methane.
I have 2 doubts:
1 - I would like to know if I have properly addressed the issue of the command “run 0” in the file “runvdwl13in.npt” to obtain the energy (van der Waals potential) of a specific configuration of 2 stationary methane molecules.
2 - Is there any way to see how the command “thermo_style custom evdwl” calculated the value provided in the log file?
Sorry in advance for the long email or if I asked any unworkable (or very specific) question.
Thanks for your time.
Best regards,
Emerson Parazzi Lyra
PhD candidate
School of Chemical Engineering
University of Campinas
500 Albert Einstein Ave, Campinas, SP, 13083-852, Brazil
Email: [email protected]
Orcid ID: https://orcid.org/0000-0002-7969-3764
system13.data.txt (869 Bytes)
logvdwl.system.data.txt_13 (2.35 KB)
runvdwl13in.npt (532 Bytes)