Dear All,
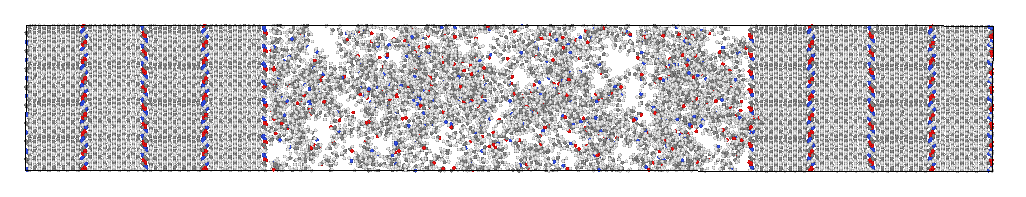
I am working on MD simulations of semi-crystal Polyamide12, using ReaxFF. I created my input geometry using Materials Studio and converted to lammps input file via msi2lmp. Here is one example:
My goal is to do deformation simulations but first I am trying to relax the system in the NPT ensemble to get rid of the internal pressure. The simulation consists of 5 stages: (1) Minimization, (2) NVE/Limit at 450K, (3) NPT at 450K, (4) NPT 450 > 300K and (5) NPT at 300K.
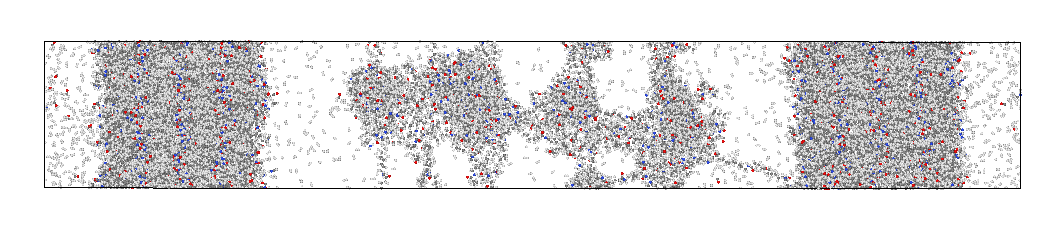
After NPT stages, I am observing some unexpected phenomenon in the resulting configuration:
This is the case after either of the NPT stages. I experimented a lot with the barostat and thermostat parameters but nothing seems to help fix the problem. Also the density of my system converges but the pressure fluctuation amplitude is quite high (it fluctuates around 0).
Has anyone experienced a problem like this? I would appreciate any help or comments.
Best regards,
Caglar Tamur
I don’t think this is caused by the barostat or thermostat settings.
It looks more like a force field issue. The use of msi2lmp rings an alarm bell. What force field did you convert the data file for? Did you edit the file afterwards?
What ReaxFF parameter file do you use? Has it been parameterized for the kind of system you are simulating and the conditions that you are simulating under?
There are a gazillion of discussions about pressure fluctuation from past mailing list posts in the mailing list archives, so I suggest you read up about this there (google will help).
Dear Dr. Kohlmeyer,
Thank you for your reply. I did the conversion using cvff and manually edited the data file afterwards; I basically deleted all the information other than masses and atoms.
I got the ReaxFF parameters directly from the author (Dr. van Duin), which was parameterized for CHON based polymer systems so I thought it would be suitable in my case.
Also this does not happen if I do fixed volume simulations like NVE or NVT, but then, I want to reduce the pressure so it seems like there is no other option than NPT?
Regards,
Caglar Tamur
I would try to first equilibrate the system using the cvff force field (or similar) and see if you get the same or different behavior.
If that works well, then I would convert to reaxff and then observe if things go bad again.
Dear Dr Tee,
I only have some XRD data and I am adjusting the crystallinity according to that. For density, I am referencing experimental results from the literature. Thank you for your comments.
Regards,
Caglar