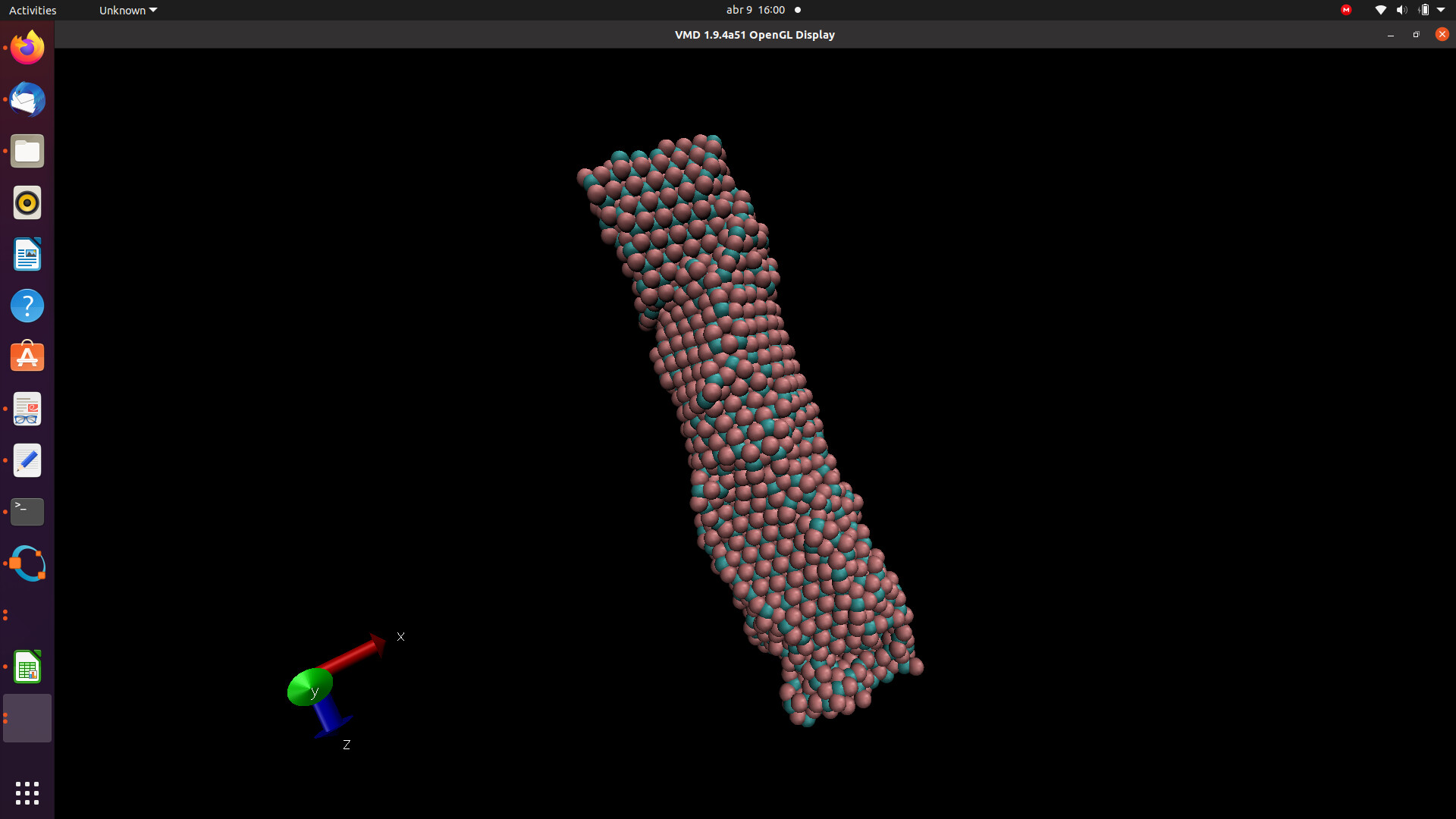
Dear LAMMPS Community,
I am trying to simulate metal oxide nanotubes with infinite length in z dimension, however I have problem with defining the size of the box in z dimension. If I use the dimensions of the structure to define z, I get some tubes folded on itself, since in the minimization stage the positions of the atoms are adjusted. Is there a command in LAMMPS that allows the simulation box to automatically adjust for an infinite periodic condition in z dimension?
Sincerely
Eduardo