Dear LAMMPS users,
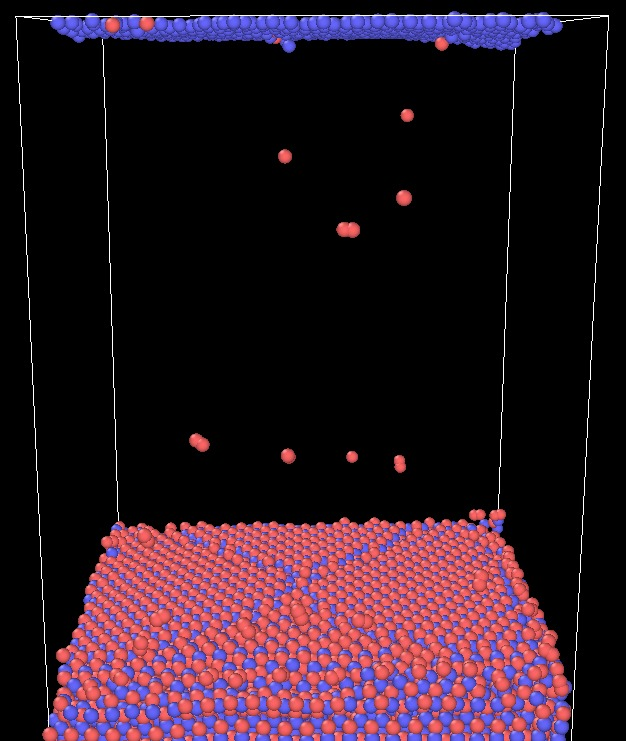
I’m trying to measure the surface energy of oxygen facet (0001) of the ZnO. But, whenever I’m running an equilibration at 298 K, O atoms at the facet just combine with each other forming O2 and evaporate out of the surface towards the Zn facet. I’ve observed this even increasing the vacuum distance from 20 to 100 A.
Does someone have experience on how to avoid such an effect?
Would it make sense to not use periodic boundary conditions only on the z axis? This way there would not be an overall electric field driving O2 to the Zn facet.
REAX potential for Zn/O/Cl system
…basal O-plane [0001]
— Initialize Simulation
clear
units real
dimension 3
boundary p p p
atom_style charge
— Create Atoms
read_data excel_surface_O_0001.xyz
— Force Field
pair_style reax/c lmp_control
pair_coeff * * ReaxFF_Potential_ZnO_Cl O Zn Cl
neighbor 2 bin
neigh_modify every 10 delay 0 check no
fix 1 all qeq/reax 1 0.0 10.0 1e-6 param.qeq
— Define Settings
compute csym all centro/atom 12
compute eng all pe/atom
compute eatoms all reduce sum c_eng
---------- Run Minimization ---------------------
reset_timestep 0
timestep 0.001
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump coord1 all custom 400 min1_Surface_O_0001.custom id x y z
fix 1 all qeq/reax 1 0.0 10.0 1e-6 reax/c
min_style cg
minimize 1e-15 1e-15 5000 5000
#undump 1
undump coord1
---------- Run Minimization 2---------------------
Now allow the box to expand/contract perpendicular to the grain boundary
reset_timestep 0
thermo 10
fix 2 all box/relax x 0 y 0 z 0 vmax 0.001
dump coord2 all custom 400 min2_Surface_O_0001.custom id type x y z
min_style cg
minimize 1e-15 1e-15 5000 5000
undump coord2
unfix 2
----------- Equilibration ---------------------------
reset_timestep 0
timestep 0.25
fix 3 all npt temp 298.0 298.0 100.0 aniso 1.0 1.0 1000.0
thermo 500
thermo_style custom step temp density press vol lx ly lz c_eatoms
dump 5 all atom 1000 dump.Surface_O_0001.ZnO
run 40000
Thank you in advance!
Best,
Gustavo