i don't have the time to discuss all details, but there are
a number of strange things going on.
why do you use s s s boundary conditions?
this way you simulate a "cluster" or "block" not a "wire"
(which to me implies using periodic boundaries in one
direction).
why do you use such a overly small time step?
with atoms as heavy as silicon, this should not be needed.
why do you use such a large drag value on the thermostat?
in order to keep the temperature at the desired range, it is
better to reduce the time constant.
also, your system is still equilibrating, that is for the initial
part of a trajectory typically better handled by the langevin
thermostat.
as for the "jump" there is not a "jump" but rather a
gradual increase which then gets a bit larger over time.
that is a sign, that your system is not yet equilibrated
and while it is continuing to go down in potential energy,
your kinetic energy goes up until at some point you have
a signficant reordering after which the structure seems
stable.
this would probably be the kind of reconstruction
that you want to see. the difference between sw
and tersoff, i likely due to the fact that tersoff has
a different (and more rugged) potential landscape,
so you may either "trapped" in a local minimum
or the reconstruction might have already happened
during the equilibrium stage.
basically, for as long as the potential energy is decreasing,
you have to keep running with nvt, or else your temperature
will go up. also, you have to remember that equilibration will
take longer at lower temperatures.
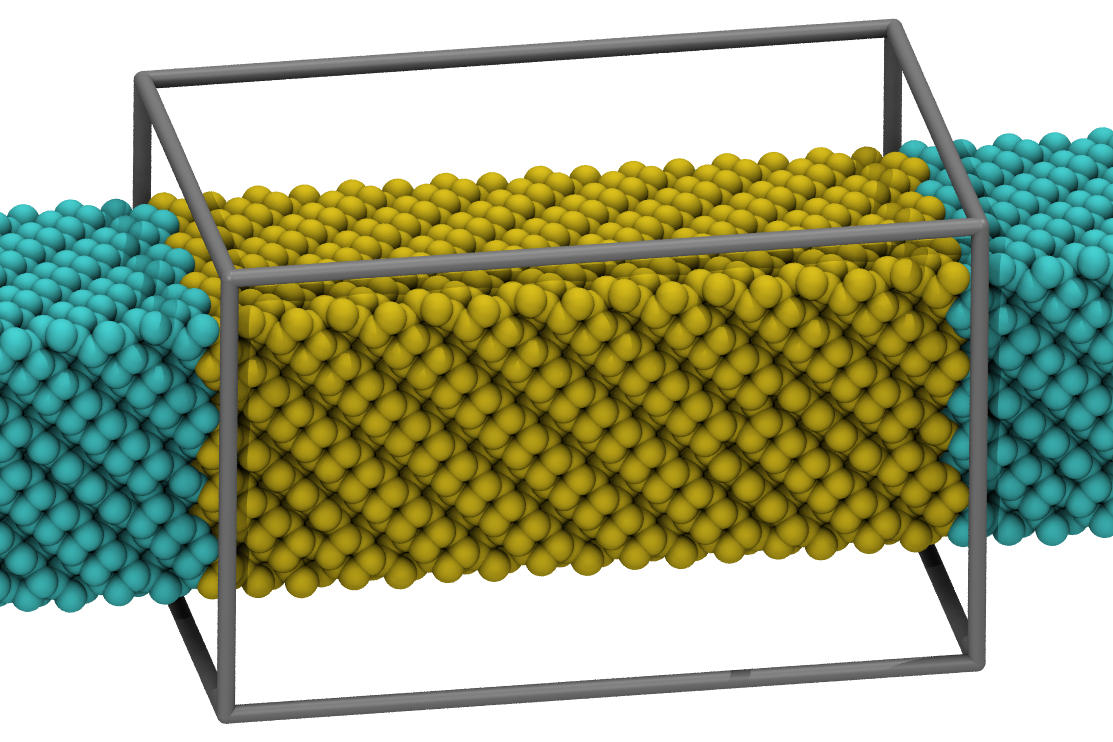
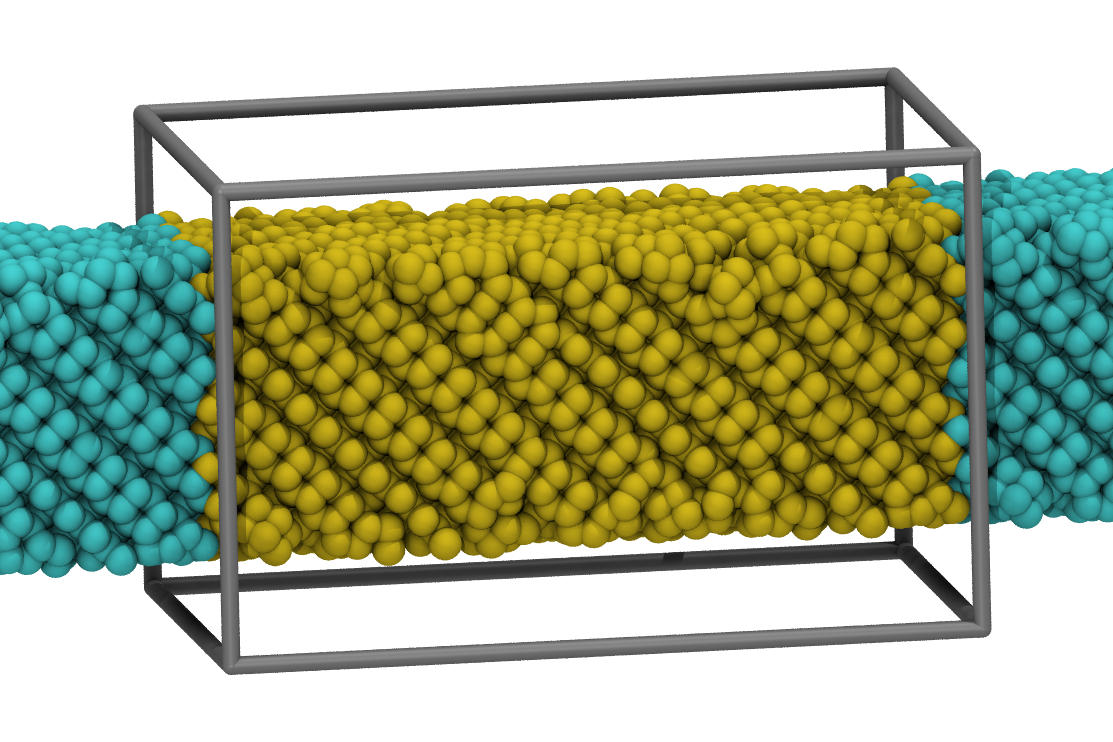
please see below how i would set up a "nanowire" simulation
with sw (or tersoff for that matter).
the system is not completely equilibrated after 2ns, but fairly
close, and you won't see any "jumps" anymore. if you replace
sw with tersoff, you should get a very similar result. only, that
the functional form of the tersoff potential also supports sp3 to
sp2 transitions, so there are some small differences in details
of the reconstruction at the edges. check out the attached
visualizations. there are many more details to consider, but i
hope this demonstrates that it is not only the method that
makes a difference, but how you go about to get what you are
after, and that MD is not a fire-and-forget tool.
axel.