Hello,
I am using the solid liquid interface method to calculate the melting point of LiAlO2 (melting temperature is roughly 2000K). I am taking a long cuboidal simulation cell of LiAlO2 and melting a small slab section in the middle of the long box to 6000 K while the rest of the box is at 1800 K. My hypothesis is that the solid liquid interface (SLI) SLI1 and SLI2 should move closer to each other with time as the rest of the box is at 1800 K which is below the melting point of LiAlO2 (MP is 2000K). size of box: 31 X 31 X 113 angstrom
Potential used: core shell potential from "
“Atomistic simulations of the defect chemistry and self-diffusion of Li-ion in LiAlO2.” Energies 12, no. 15 (2019): 2895.
density and structure are correctly reproduced from these potentials.
The inputs and output are given below
Z direction --------->
SLI1 | SLI2 | |
solid | liquid | solid |
1800 K | 6000K | 1800K || |____________ |
The code is given below:
units metal
dimension 3
boundary p p p
atom_style full
fix csinfo all property/atom i_CSID
read_data largecell22.lmp fix csinfo NULL CS-Info
group cores type 1 2 3
group shells type 4
variable z14 equal 0.40zhi+0.60zlo
variable z34 equal 0.60zhi+0.25zlo
region midregion block INF INF INF INF {z14} {z34}
group midgroup region midregion
fix zeromomentum all momentum 1 linear 1 1 1
variable myT equal 1800
velocity all create ${myT} 87287
velocity all zero linear
pair_style buck/coul/long/cs 10 12
pair_coeff * * 0.0 1.000 0.00
pair_coeff 1 4 632.1018 0.2906 0.00 #Li core O shell
pair_coeff 2 4 1109.92381 0.31540 0.00 #Al core O shell
pair_coeff 4 4 12420.5 0.2215 29.07 #O shell O shell
bond_style harmonic
bond_coeff 1 31.0 0.0
kspace_style ewald 2.0e-3
neighbor 1.0 bin
neigh_modify every 20 delay 0 check no
variable time equal step*dt
thermo 100
thermo_style custom step v_time temp pe ke etotal vol press
thermo_modify norm no
initially heat up to ${myT} K, will be reached after about 10 ps with nosehoover npt
to get the correct volume
fix 1 all npt temp {myT} {myT} 0.1 aniso 0.0 0.0 0.1
dump dump1 all custom 1000 dump.therm.gz id type x y z
#dump_modify dump1 sort id
timestep 1.0e-3
run 10000
#write_restart restart.therm
#undump dump1
unfix 1
over melt the mid with berendsen 50 ps
dump dump1 all custom 1000 dump.overmeltmid.gz id type x y z
#dump_modify dump1 sort id
reset_timestep 0
fix 1 midgroup nve
fix 2 midgroup temp/berendsen 6000.0 6000.0 0.1
run 50000
#write_restart restart.overmeltmid
#undump dump1
unfix 1
unfix 2
compute K all ke/atom
variable keeV atom “c_K * 0.043364102”
velocity midgroup create {myT} 87287
velocity all zero linear
dump dump1 all custom 1 dump.melt.gz id type x y z vx vy vz fx fy fz v_keeV
dump_modify dump1 sort id
reset_timestep 0
fix 1 all nve
fix 2 all temp/berendsen {myT} ${myT} 0.1
run 250000
#write_restart restart.melt
undump dump1
unfix 1
unfix 2
Output:
I am plotting the kinetic energy (representative of temperature) vs z coordinates at 2 time instances, First at the beginning of the simulation and second after 250 steps. My time step is 0.001 ps. I expect to see the peak of the kinetic energy to become narrower signifying that the SLIs are moving closer to each other. But instead I am seeing that peaks flattens and broadens.
Question 1: Is my approach correct? Is there a better way to quantitatively track the movement of solid liquid interface?
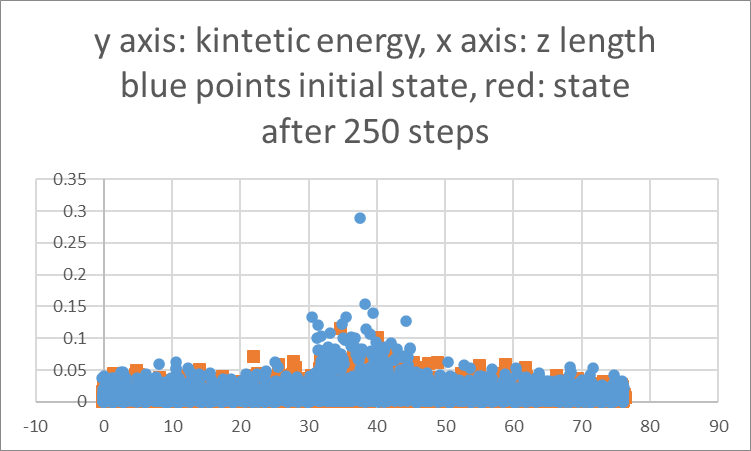
Question 2: If I raise the temperatures of the side sections to 2500K can I expect the SLIs to move away from each other?
The observation for T= 2500K is shown below.
I am not able to see the SLIs move away because the red peak is not becoming wider. Is my hypothesis wrong?
Why are there high energy points at the boundaries in both initial and final states?

Sincerely,
Ankit