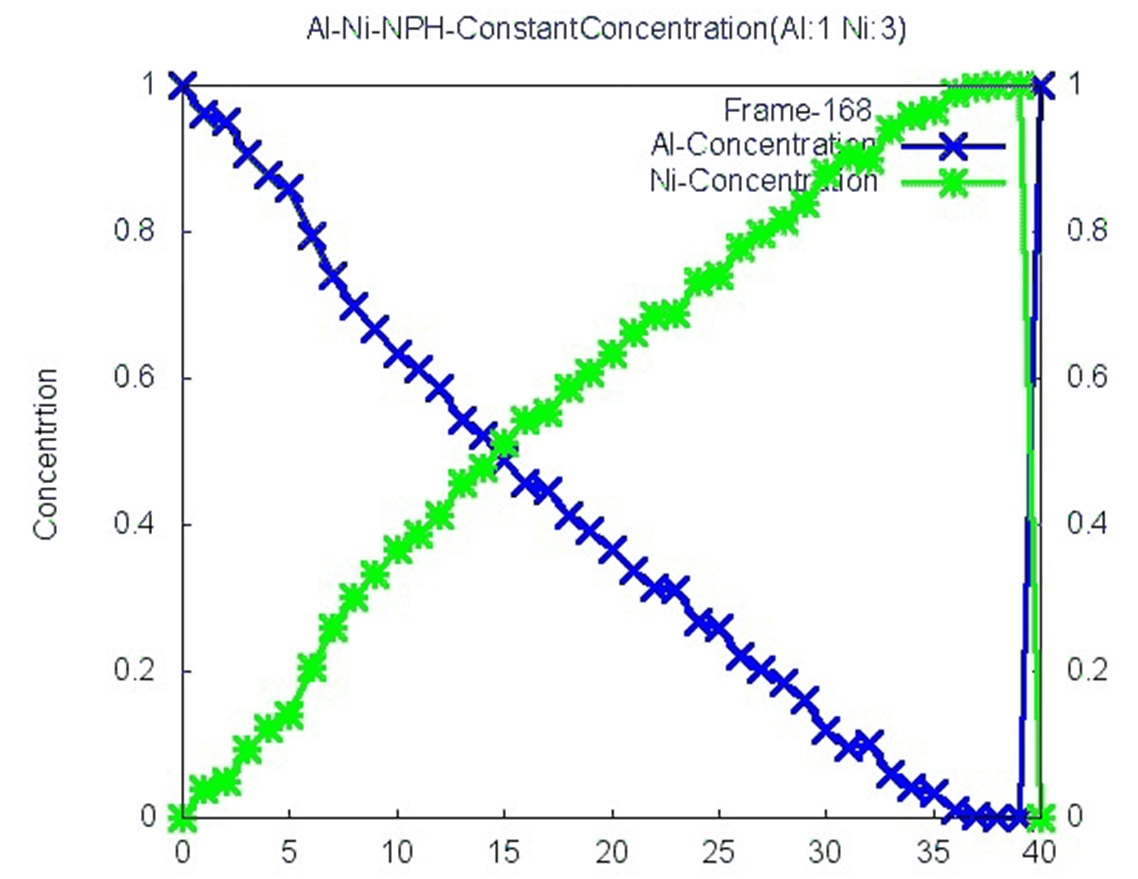
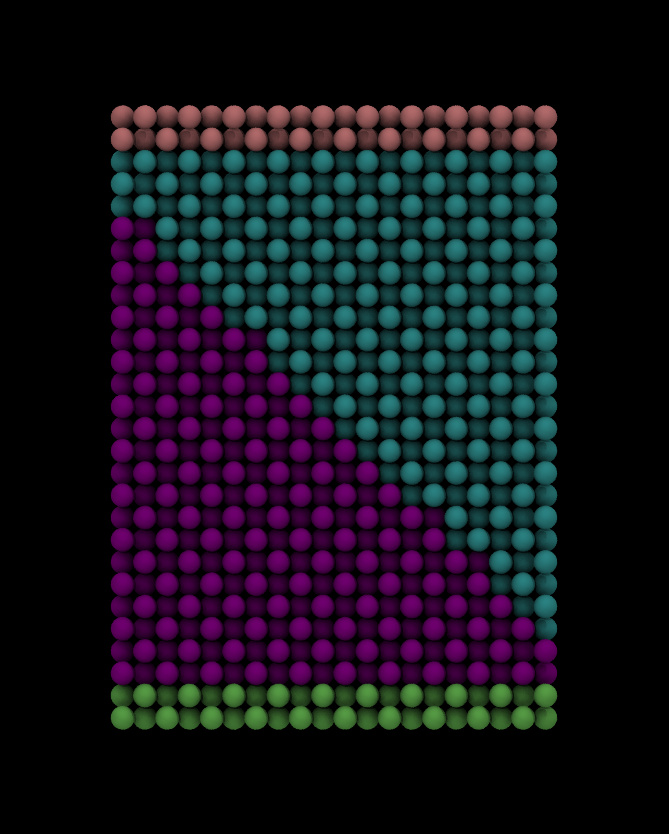
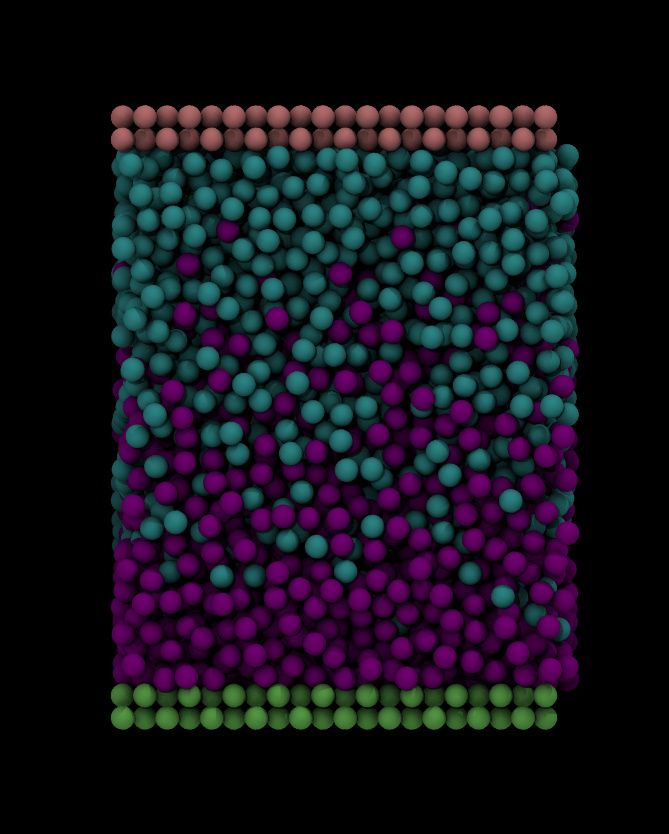
I need to do that because eventually i want to heat the system up to 1750K
and hope to establish a constant concentration gradient within the system.
If i dont "reset the atom types of those atoms at the two boundaries
periodically", ill just get random Al-Ni mixing. I attached a figure to this
email, it can probably help in explaining my goal.
if it is just about the concentration gradient, why don't you just
start from two wedges of both atom types and then modify
fix spring/self so that the restoring force is only applied in z-direction.
this way the concentration gradient is predefined by your initial
conditions and cannot change.
you'll have to explain what you need that gradient for.
Basically im "reassigning groups and resetting atom types" periodically to
maintain a 100% boundary condition. I am try to use the "if" command to do
this, and i wonder if "mod" works in "if"? Can i do something like "if
"${steps} mod 10000"==0 then blah blah blah"?
i don't know, but there a several serious concerns about that approach.
worst of all. you are changing atom types for elements that are very
different in size and mass. that will have two (unphysical) effects.
due to the difference in mass the individual kinetic energy of the
atoms will change by about a factor of two, i.e. ni atoms that were
al before, will suddenly be twice as "hot" and new al atoms only half.
second, due to the difference in size, you will be generating small
shock waves that will keep bouncing back and forth through your
system, the excess kinetic energy due to that will have to be
constantly removed by the thermostat. i cannot imagine that you
can compute any property of such a system without it being
significantly tainted by those effects.
As for the dimensions, i calculated the size of one Al atom layer and one Ni
atom layer, i dont really understand what u mean by equillibration, but ill
try to fix anything u point out.
you start from a perfect crystal and then run with nvt and then npt.
during the npt run, your system will change size and atoms may
move around. at least in x and y you can pick much larger dimensions
to make sure you catch all atoms. in z-direction you have to hope
that the system doesn't deform or drift too much.
also, i am not at all convinced that your scheme will actually
result in that kind of gradient that you are plotting (how did you
create that plot?).
So do you see what im trying to do with "resetting atom types" periodically
now?
you are putting yourself in great misery? 
but seriously, you are only describing the immediate problem,
not the "big picture". if you explain what the purpose of building
that concentration gradient is supposed to be and what kind of
support you have that your method will be suitable to achieve it,
you may get better help.
axel.