Hello Steve,
Thanks for your reply!
In fact, I have made a new test about this problem. More results can be found in the e-mail (2021-05-13) “Two problems when we use the nvt/sllod command in Lammps.” and the latter reply.
I make it shortly.
(1) I used the Kremer-Grest model, whose the data file can be found in BENCH folder in lammps (32000 monomers, chain length is 100).
(2) Before I imposed the shear, I clear the velocity of center-of mass (“velocity all zero linear”) and changed the box to be triclinic (“change_box all triclinic”).
(3) Then, I imposed the shear by “fix nvt/sllod” with “fix deform”:
fix 1 all nvt/sllod temp 1.0 1.0 1.0 tchain 1
fix 2 all deform 1 xy erate 0.001 units box remap v
Note: shear rate is 0.001
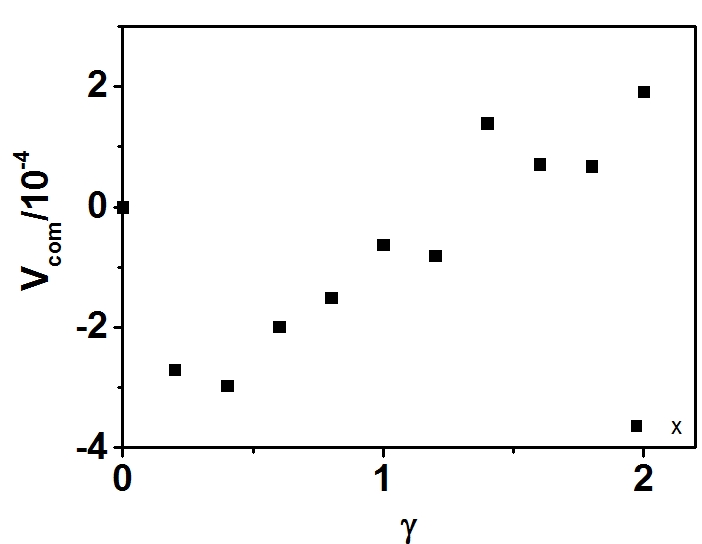
First, I have shown that the velocity of center of mass increases with the strain. In other words, the velocity of center of mass is not a constant.
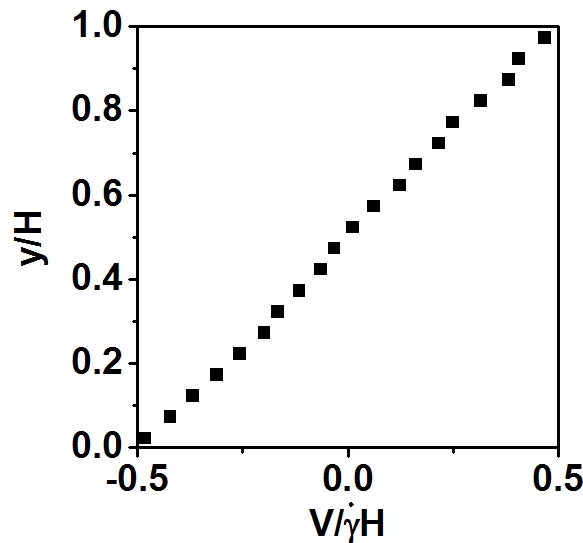
Second, Axel tell me using the following command to get the velocity profile:
compute ybins all chunk/atom bin/1d y lower 0.05 units reduced
fix 5 all ave/chunk 40 5000 200000 ybins vx file yprof.dat
The results is shown in below, which states the velocity is range from -V/2 to V/2. Thus, the COM velocity should be zero, not V_max/2.
Third, I read the fix deform.cpp as possible as I can (I am much more familiar with Fortran). I think if the box is [0, H_y],we can get “atoms at the lower face will be (near) zero velocity. Atoms at the upper face will have a large
velocity.” However, my box is [-H_y/2, H_y/2]
If possible, you can tell me what I can do, I will try my best to check it.
Sincererly,Yongjin Ruan