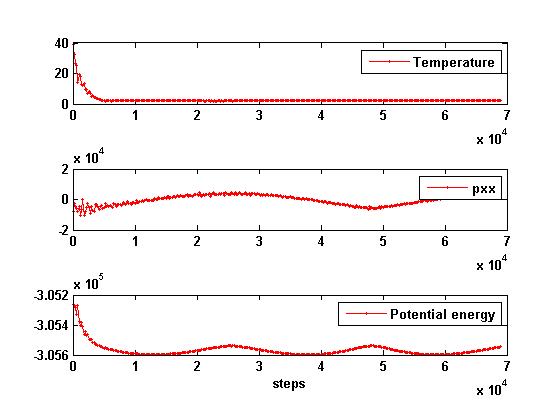
I am simulating a Au nanowire build from bulk lattice constants.
I want to relax the system to its equilibrium.
The pressure a long the transverse direction is ok.
However, the pressure in the length direction and the potential energy of the system oscillate a lot
and do not seem to converge to a stable value, as shown in the attachment.
The following is the main code I have used.
Pressures oscillate. It's the nature of MD. If you
start with free surfaces that are unrelaxed, and let
them go, you may well get oscillations. It's like
releasing a stretched spring and there's nothing
to damp the motion.
Thank you.
Is there any possible approach to get rid of the oscillation?
Moreover, if the nanowire is loaded with a external force in the tranverse
directions,it will also oscillate.
It is quite difficult if I want to average the potential energy and the
displacement.
Again, this is more of a MD issue, than a LAMMPS issue.
You can run a bigger system. You can pre-equilibrate free
surfaces with a minimize command, to reduce oscillations
during dynamics. You can damp the motion a few times,
then you should be able to run NVE w/out big osciallations.
But if you are driving the system with a boundary condition
then all bets are off.