Dear all,
I have noticed a possibly wrong example given in the documentation page of “lattice”, regarding the Wurtzite lattice structure. There has been a thread raising this issue before, but it was never justified or explained what was the problem exactly and the thread stopped.
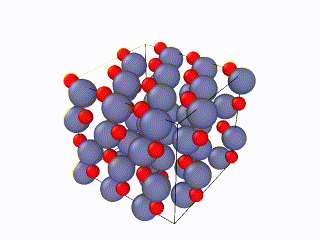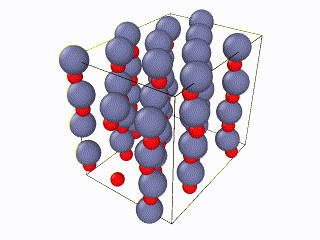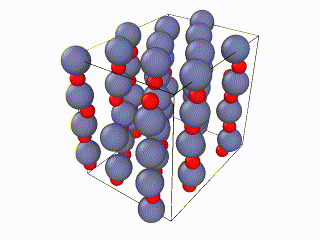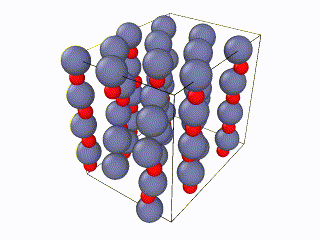
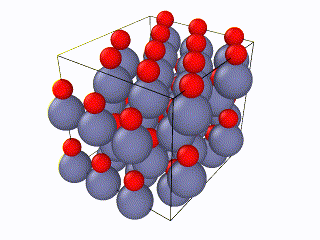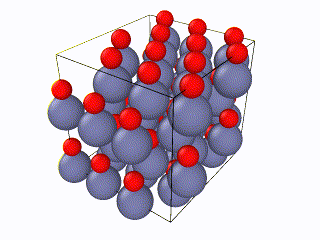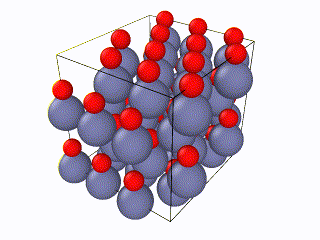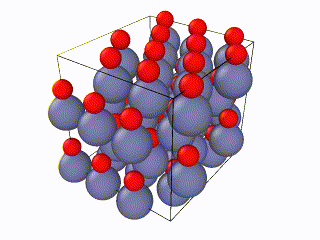
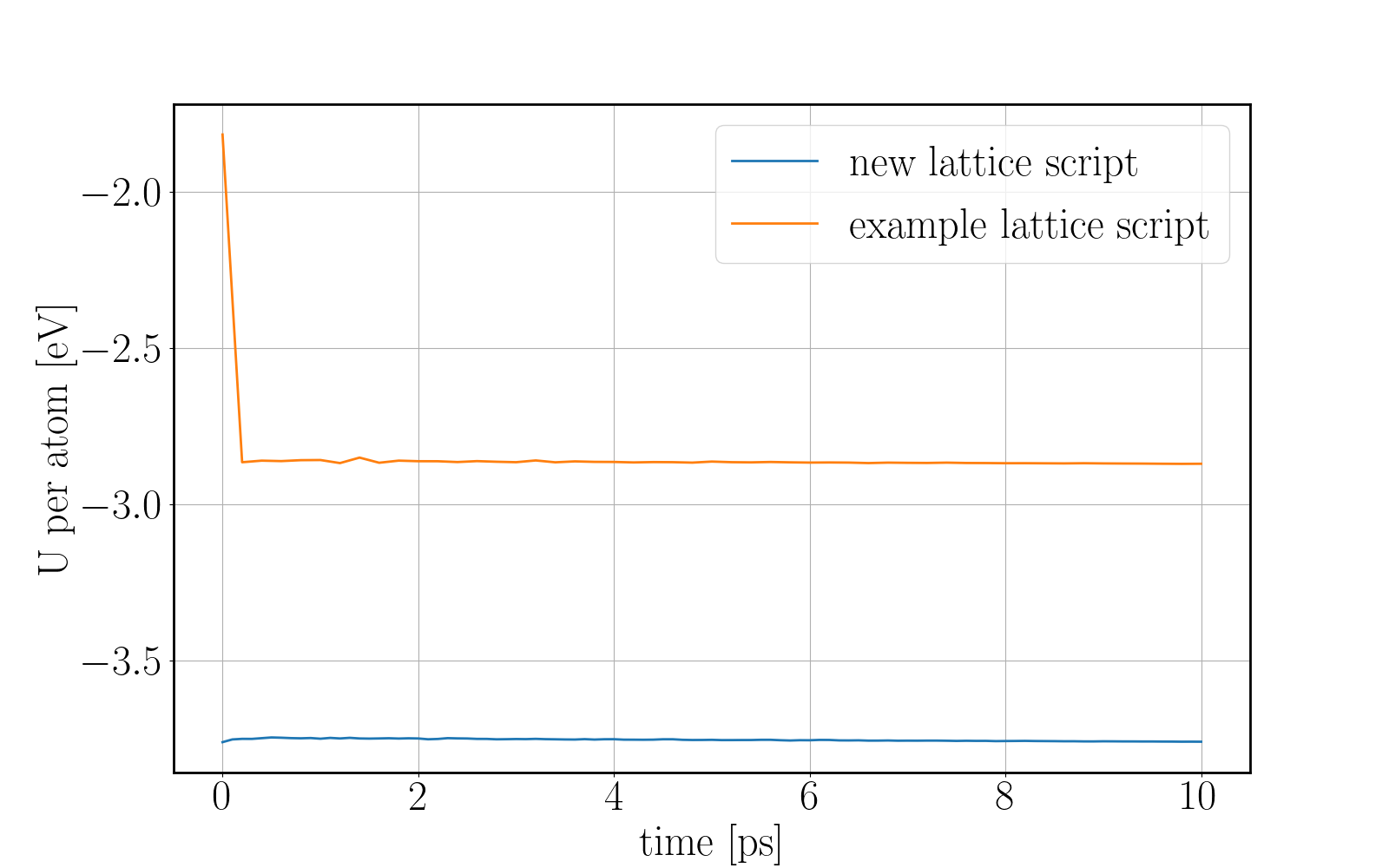
I have been trying to run some simulations for Wurtzite ZnO. Initially I created the lattice by using the example script from the documentation file and I ran into problems. When relaxing the structure, I saw an immediate restructuring, with the system starting from quite high energy values and rearranging significantly. All simulations were ran using the Tersoff potential by Erhart et. al., which is considered quite reliable for ZnO. The relaxation input script is attached as “wurtzite_ZnO_ldoc.in” As you can see in the attached gif, there is significant restructuring and energy reduction. The initial structure was not relaxed at all which can be clearly seen both in the evolution movie:
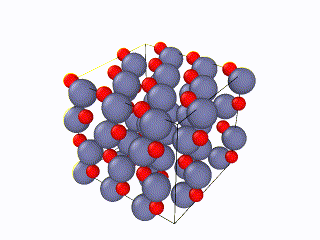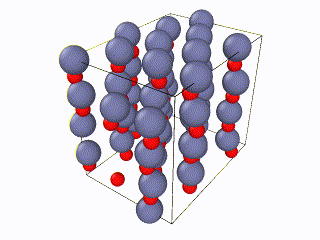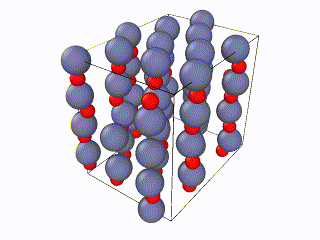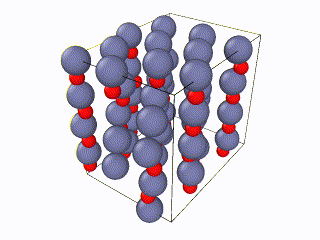
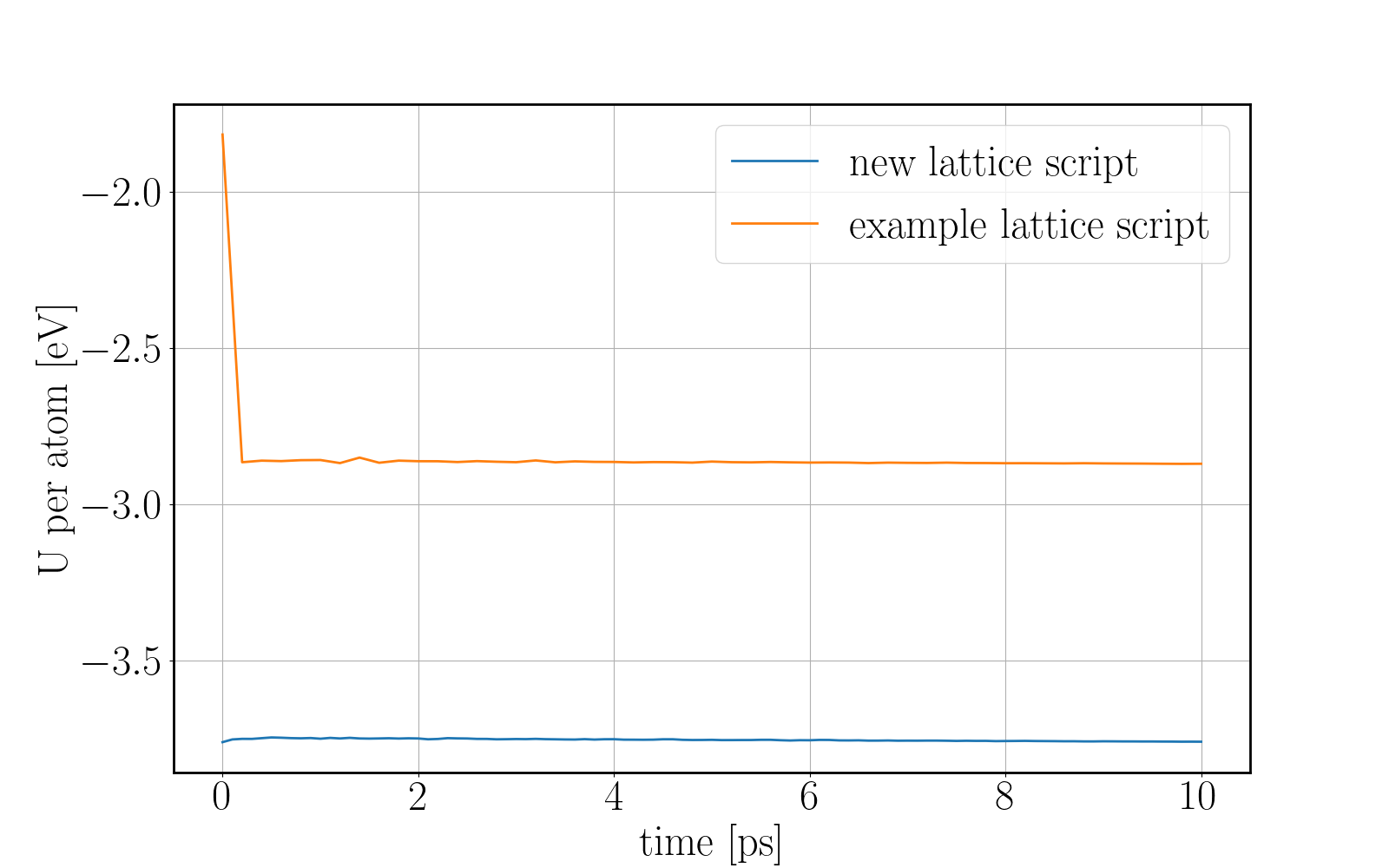
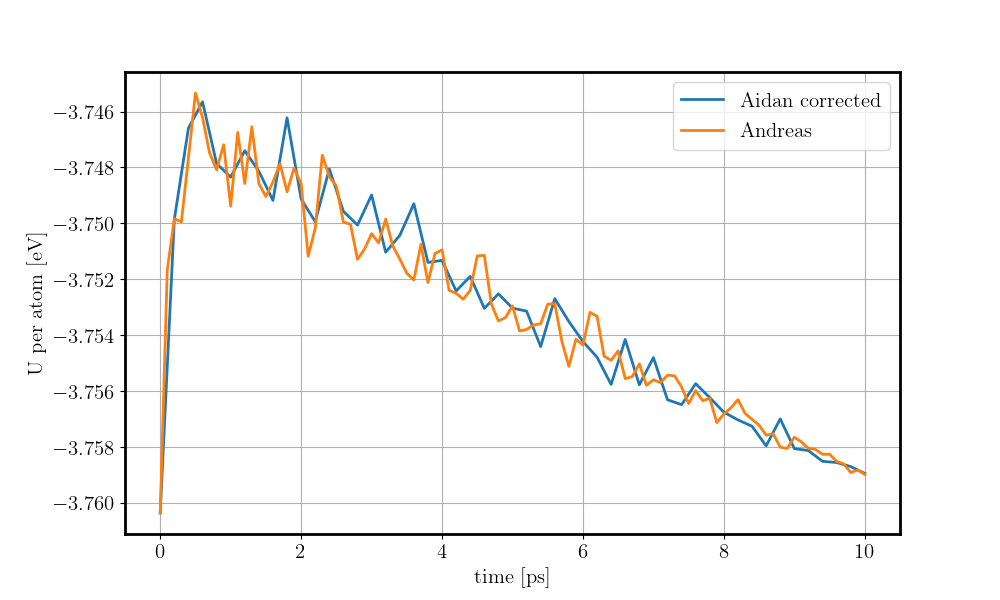
and the energy reduction at the energy plot.
I tried to find what exactly the error is, but I couldn’t. I think something is wrong with the box periodic boundaries, but I am not sure. In any case, I created my own lammps script for the Wurtzite structure, using the primitive unit cell as described here. I ran it with exactly the same relaxation script (attached as “wurtzite_ZnO.in” ) and it seems to be working perfectly and be well-relaxed. Here is the corresponding gif:
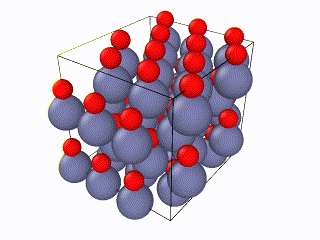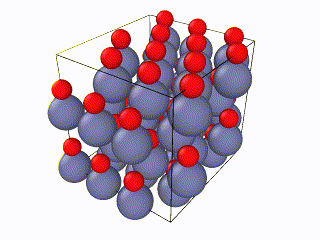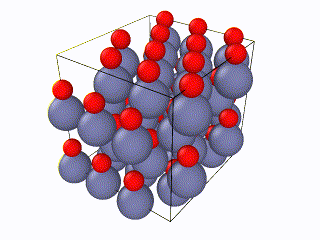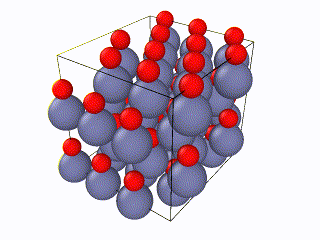
Here you can see a comparison of the potential energy evolution of the two crystals, showing that the initial lattice created from the example script is not relaxed wurtzite ZnO.
I suggest that the example script is corrected in the documentation page, or possibly replaced by the one I propose (attached).
Best regards,
Dr. Andreas Kyritsakis
wurtzite_ZnO.in (2.04 KB)
wurtzite_ZnO_ldoc.in (1.5 KB)