Dear Sir,
I am writing a code to calculate lattice parameter of BCC Fe 5% Ni. I am deleting a spherical region out of rectangular box and putting Ni atoms into the deleted space. Then I am minimizing my system for Ni to distribute randomly in the simulation block.
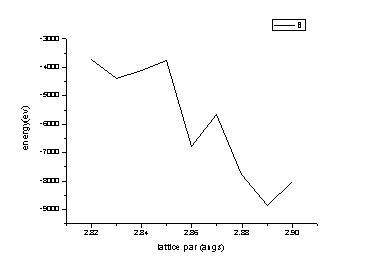
Potential energy is always found ta have abrupt values with changing lattice parameters, rather it should be minimum at about 2.868-2.869 angs.
Please help me in knowing where I am going wrong.
Agraj
Attached here with is the *.jpg file of energy values at various lattice spacing.
log lattice3.lammps
variable y equal 2.70
label iterations
atom_style atomic
units metal
boundary p p p
neighbor 2.0 bin
neigh_modify check yes
lattice bcc $y
region box block -6 6 -6 6 -6 6 units lattice
create_box 2 box
create_atoms 1 box
region sphere sphere 0.0 0.0 0.0 3.55 units lattice
delete_atoms region sphere
lattice fcc 3.52
create_atoms 2 region sphere
group Fe type 1
group Ni type 2
mass 1 56
mass 2 58
atom_modify sort 1 2.0
fix 1 all nve
run 500
pair_style eam/alloy # Uses Embedded atom method - to compute interactions
pair_coeff * * Fe-Ni.eam.alloy Fe Ni
min_style cg
minimize 10e-4 10e-4 10000 10000
variable e equal pe # Defines variable e as potential energy
run 500
print “The current lattice parameter value is $y” # Prints current lattice parameter
print “The current potential energy value is $e” # Prints potential energy
variable y equal $y+0.01 # y = y + 0.1
clear
if $y <= 3.0 then “jump latticeparameter.txt iterations”
Dear Sir,
I am writing a code to calculate lattice parameter of BCC Fe 5% Ni. I am
deleting a spherical region out of rectangular box and putting Ni atoms into
the deleted space. Then I am minimizing my system for Ni to distribute
randomly in the simulation block.
that is not going to happen. if you want particles to be scattered
randomly, you have to do that at random positions. if you are picking
random positions, like in any monte carlo scheme, you'll have to
average over a large enough number of samples, and you have
to come us with some MC particle type swap plus relax scheme
to find an equilibrium.
Potential energy is always found ta have abrupt values with changing lattice
parameters, rather it should be minimum at about 2.868-2.869 angs.
Please help me in knowing where I am going wrong.
GIGO.
axel.
Just to add...
If you do go the MC eq route, you might want to check out: Foiles, S.M., Physical Review B 40 (17), 11502-11506 (1989).
*For your system, it may be difficult to obtain reasonable acceptance probability if the Ni is occupying interstitial positions. (swapping a Ni atom with a vacancy)
Axel Kohlmeyer wrote:
Then I am minimizing my system for Ni to distribute randomly in the simulation block.
If I understand what you are doing, you have a Fe crystal, you cut out
a sphere, you
put a lattice of Ni atoms in the spherical void, then minimize. That is not
going to distribute Ni randomly throughout the Fe. You would see that
if you visualized the result. I think LAMMPS did what you asked it.
You could use the set type/fraction command to change the type of a
random set of atoms to Ni from Fe. All on one lattice.
Steve
Dear Dr. Axel, Mr. Michael thanks for suggestion as I am not aquainted with Monte Carlo, I tried to go for type//fraction command only.
Sir, As per your suggestion I have introduced type/fraction command for replacing Fe by Ni and Cu. I am calculating lattice parameter at 400 deg C. But still I am not getting “U” shaped variation of potential energy as a funtion of lattice parameter. Potential energy is slightly fluctuating up and down (just like Serration) near the desired lattice constant (2.85-2.87).
I fear if my code is correct or not, or potential is designed to calculate lattice parameter and other properties for Fe30Cu65Ni5 ternary alloy system.
Agraj
#INPUT FILE#
log Fe30Cu65Ni5.lammps
variable y equal 2.6
label iterations
atom_style atomic
units metal
boundary p p p
lattice bcc $y
region box block 0 10 0 10 0 10
create_box 3 box
create_atoms 1 box
set group all type/fraction 2 0.65 25462
set group all type/fraction 3 0.05 12342
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Fe Cu Ni
min_style cg
minimize 10e-4 10e-4 5000 5000
fix 1 all npt temp 673 673 0.1 iso 0.0 0.0 1.0 drag 1.5
fix 2 all temp/rescale 1 673 673 1 1
run 500
variable e equal pe
thermo_style custom step temp pe ke etotal press pxx vol
thermo 100
run 1000
print “The current lattice parameter value is $y”
print “The current potential energy value is $e”
variable y equal $y+0.01
clear
if “$y <= 3.0” then “jump latpara iterations”
Dear Dr. Axel, Mr. Michael thanks for suggestion as I am not aquainted with
Monte Carlo, I tried to go for type//fraction command only.
Sir, As per your suggestion I have introduced type/fraction command for
replacing Fe by Ni and Cu. I am calculating lattice parameter at 400 deg C.
But still I am not getting "U" shaped variation of potential energy as a
funtion of lattice parameter. Potential energy is slightly fluctuating up
and down (just like Serration) near the desired lattice constant
(2.85-2.87).
I fear if my code is correct or not, or potential is designed to calculate
lattice parameter and other properties for Fe30Cu65Ni5 ternary alloy
system.
it is not the code, it is the way you are addressing the problem.
a) if you run at finite temperature, you have to first equilibrate and then
average over a long enough time and take that average as input,
not the instantaneous energy as that is fluctuating hence the noise
b) you have to check, whether your (pairwise) potential energy is shifted
to 0 near the cutoff. otherwise when using a fixed cutoff but are varying
the lattice parameter, then you'll have jumps in energy due to atoms
suddenly being no longer within range of the cutoff
c) have a look at how stat mech text books address this issue, e.g.
fit your data to a suitable equation of state and then compute the minimum
from that fit.
all three issues mean, that you'll probably be better off doing a
little extra "home work" in the library before getting back to running
simulations on the computer. research is rarely as straightforward
as it may appear at first.
cheers,
axel.