I’m currently working on my final project which is working on functionalized graphene with OH.
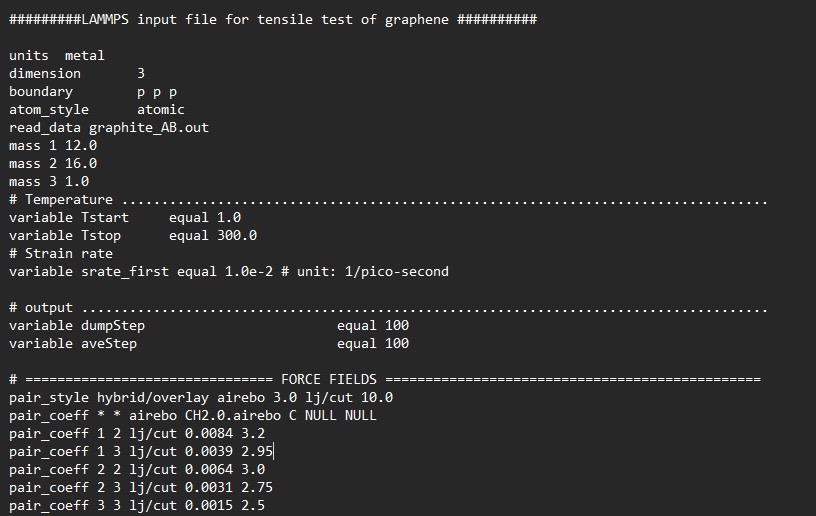
My question is regarding the force field I have used Airebo to define the interaction between the C-C.For others interactions, I used LJ potential the results I got when I run the simulation ( OVITO ) doesn’t make sense and It shows instant break before running the test.do I have to mention other parameters or Sigma and epsilon of the interactions should be sufficient to allow me observe the interaction between the graphene and the OH
Simulations strictly follow the GI-GO principle (= garbage in, garbage out).
Do you have any evidence that you can model graphene functionalized with OH groups by only using LJ interactions? I would be very surprised if that would work.
Traditional molecular force fields also need bonds and angles and partial charges assigned to those atoms. Which also means that you cannot run with atom style atomic.
You will need to do some more research into what would be a suitable force field and what set of parameters for that specific force field would be suitable for your material and the conditions that you are simulating. Please keep in mind that for classical models, it is not sufficient to only mention the elements (unlike in quantum chemistry), but force fields and parameter sets are quite specific and may need to be different for different compounds and conditions. I would perhaps do some research if there is a suitable ReaxFF parameter set that was testing successfully on functionalized graphene. This seems to be a popular enough topic so that some publications should exists, if it has been done and worked out.
A few more comments:
using pair style hybrid/overlay makes no sense. just hybrid should suffice for a mixed force field setup and doesn’t have the risk of double counting.
you seem to have a modified AIREBO potential file. with things like that all bets are off anyway.
From “final project” I assume you are an undergrad student – a “tensile test” in MD is a difficult simulation using an inappropriate technique.
Almost all MD “force fields” assume that each atom’s electronic structure stays almost constant throughout the simulation. This is why we can get away with harmonic spring bonds and angles, LJ repulsions, and atom-centered partial charges.
By contrast, a tensile test requires bonds to be severely deformed or even broken. The energy required for such changes goes into reorganising the electrons in those bonds, but that breaks the usual MD assumptions. Even “many-body” force fields like AIREBO, that try to dynamically impose bonding-type forces, will struggle in these more extreme scenarios.
These many-body force fields require more computational time as well as careful validation – not suitable for an undergraduate project unless you have a very involved and knowledgeable supervisor. They are typically used in more advanced research because the next “fool-proof” method, DFT, is even more time consuming. Furthermore, a many-body force field has to be carefully calibrated to a particular reaction context and particular set of atoms. For example, a “reactive force field” tuned for burning plastic including nitrogen atoms will have some assumptions (such as being better suited for gases instead of liquids) that don’t hold up in an aqueous solution with only carbon, hydrogen and oxygen.
I would strongly suggest a topic with a different focus for undergraduate research. For example there are many topics about ions at the interface between an electrode, especially charged, and the electrolyte, that have not been well-studied and are suitable for quick simulations. Note that this forum is usually for more technical discussions on how to use LAMMPS, not what to study with it, so you should discuss more with your supervisor or other local assistance to modify or change your project.