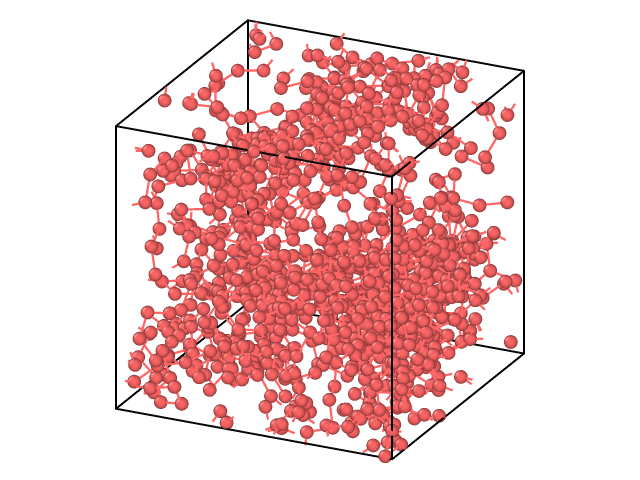
I am using a corse-grained model of PMMA where each particle represents a monomer It is the amorphous result of:
1-) Creation of fcc crystal of particles, 2-) Minimization, 3-) Equilibration in NVT at 300 K, 3-) Heating up to 1000 K, 4-) Equilibration in NVT at 1000 K, 5-) Cooling down to 300 K, 6-) Equilibration in npt using (300 K and 10 bar)
I have done all these steps using “boundary p p p”.
Now I’ve changed to “boundary p p f” in order to perform deposition. The problem is that I am losing atoms from the surface. Is there a problem? If so, is there a way to minimize it?
I am using a corse-grained model of PMMA where each particle represents a
monomer It is the amorphous result of:
1-) Creation of fcc crystal of particles, 2-) Minimization, 3-)
Equilibration in NVT at 300 K, 3-) Heating up to 1000 K, 4-) Equilibration
in NVT at 1000 K, 5-) Cooling down to 300 K, 6-) Equilibration in npt using
(300 K and 10 bar)
I have done all these steps using "boundary p p p".
Now I've changed to "boundary p p f" in order to perform deposition. The
problem is that I am losing atoms from the surface. Is there a problem? If
so, is there a way to minimize it?
you can avoid losing atoms by using an "m" or "s" boundary or adding a
wall potential.
if you want to deposit additional atoms, you also need a volume to deposit
atoms into and for that you have to move the box boundaries, too, which
only works with "f" or "m" boundary.
Now, I am using a differente model for PMMA where each particle represents a monomer, but the monomers are connected now by FENE bonds.
I did the equilibration using “boundary p p p” and everything was ok.
Now I need to perform Au deposition on its surface. I have put a wall potential (wall/lj93) on the z high and I am using z boundary m. I got the message:
WARNING: Bond/angle/dihedral extent > half of periodic box length (…/domain.cpp:895)
WARNING: FENE bond too long: 1000 27 28 108.439 (…/bond_fene.cpp:91)
ERROR on proc 0: Bad FENE bond (…/bond_fene.cpp:92)
How can I proceed to “open” my up surface to deposition without this problems?
Now, I am using a differente model for PMMA where each particle
represents a monomer, but the monomers are connected now by FENE bonds.
I did the equilibration using "boundary p p p" and everything was ok.
Now I need to perform Au deposition on its surface. I have put a wall
potential (wall/lj93) on the z high and I am using z boundary m. I got the
message:
WARNING: Bond/angle/dihedral extent > half of periodic box length
(../domain.cpp:895)
WARNING: FENE bond too long: 1000 27 28 108.439 (../bond_fene.cpp:91)
ERROR on proc 0: Bad FENE bond (../bond_fene.cpp:92)
How can I proceed to "open" my up surface to deposition without this
problems?
your approach with first using fully periodic boundaries and then
switching to non-periodic is not a good idea. you are only equilibrating
the inner part of your system. the surfaces need significant equilibration
work after changing the boundaries.
it is better to start with p p f right away and then also place a pair of
harmonic or lj walls to confine the system during your
relax/heat/quench/relax/equilibrate process. there is no need to do NPT,
you can simply remove the walls at the last step and (at least temporarily)
switch from f to m boundary. the system will relax into the direction of
the free surface. if you still want to do NPT, you should still be able to
do so, but you have to selectively couple of x and y direction only.
I am trying to do what you suggested. Take a look at my code below, please.
I tried to make finite boundary and put the walls at this stage, when I have to solve the atoms overlapping (pmma.dat is the data just produced by chain.py).
For the p p p boundary I can solve the atoms overlapping using the soft potential + fix nve OR using fix nve/limit with hard potential, without any problem.
But with the bellow configuration I get:
WARNING: Bond/angle/dihedral extent > half of periodic box length (…/domain.cpp:895)
WARNING: FENE bond too long: 0 692 693 107.437 (…/bond_fene.cpp:91)
ERROR on proc 0: Bad FENE bond (…/bond_fene.cpp:92)
I tried also to solve the overlapping first with p p p and only after change the boundary to p p f and put the walls, bu I got the same problem. Note I am using metal units.
Am I missing any information?
PMMA FENE BONDS
units metal
atom_style bond
special_bonds fene
boundary p p f
read_data pmma.dat
mass 1 100.0
neighbor 2.0 bin
neigh_modify every 1 delay 1
I have tried also using the benchmark FENE beadspring modified as follows, and I got the same problem (with fix wall harmonic and also fix wall lj/126). I also tried some combinations of lj parameters. I read the fix wall documentation over and over and I can't realize what is the problem. I think it should be that simple.
I have tried also using the benchmark FENE beadspring modified as
follows, and I got the same problem (with fix wall harmonic and also fix
wall lj/126). I also tried some combinations of lj parameters. I read the
fix wall documentation over and over and I can't realize what is the
problem. I think it should be that simple.
you are not paying attention and you are trying to solve the wrong problem
in the wrong place in the wrong way.
the problem is due to *changing* the boundary conditions with bonds
stretching across boundaries. that *cannot* work. start with p p m right
away. i've already explained in detail why starting with p p p for the
system you want to do is a bad idea (to put it politely).
Yes, ok, I understood.
So, I have tried to start with p p m right away, with data file just produced by chain.py. But I got the same message.
But I think the problem is the way the file is produced by chain.py (I don’t mean chain.py has a problem!). I can see in ovito there are some “bad FENE bonds” at the boundaries (bad, if the boundaries are not periodic).
Yes, ok, I understood.
So, I have tried to start with p p m right away, with data file just produced by chain.py. But I got the same message.
But I think the problem is the way the file is produced by chain.py (I don’t mean chain.py has a problem!). I can see in ovito there are some “bad FENE bonds” at the boundaries (bad, if the boundaries are not periodic).
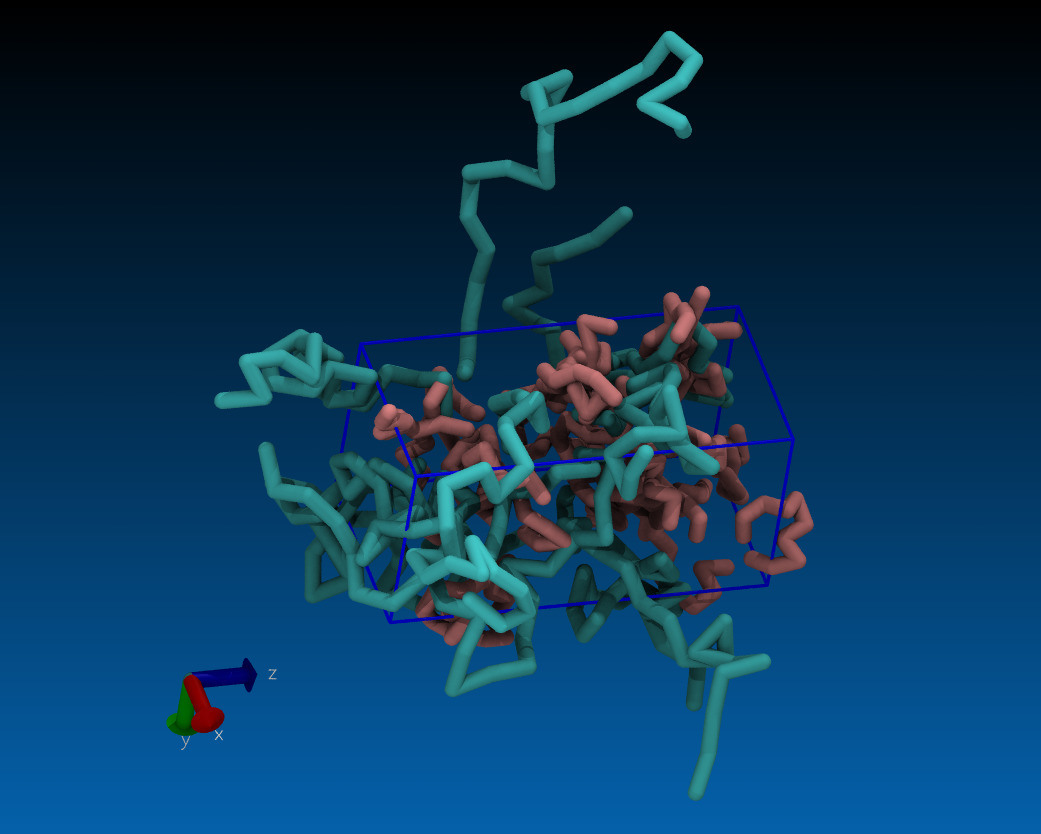
in order to put this meandering thread to a rest, i've produced a demo
example (attached).
some core issues:
- the chain tool in Pizza.py does produce proper data files with image
flags included.
- one has to be careful what the bond length parameters are. and for system
preparation purposes, it is better to stick with harmonic bonds, which
cannot break.
- since we're making changes to the system that may translate atoms over
longer distances, it is helpful to choose a long communication cutoff.
- the data files produced from the chain tool have high potential energy,
so one should start with a minimization.
- in this particular case, i scale the box twice and each time follow it
with a minimization. the purpose of this and the following MD is to create
an empty space so that PBC can be broken
- one has to check the original geometry carefully, if there is a chain
going straight through the cell, it may not be possible to "compact" it,
then one can try a different dimension or use a different RNG seed in the
chain.py script. (see the attached example visualized with VMD).
- i use here a combination of fix addforce and fix oneway to push atoms
toward the middle of the elongated periodic box. in principle, one can use
fix oneway for both, or fix addforce, but in this case the combination
seems to work best.
- look at the visualization of the trajectory, how there is a quickly
emerging vacuum in the cell
- after that it is possible to switch from periodic to non-periodic without
a problem. also it is possible to switch from harmonic bonds to fene and
reduce the communication cutoff (possible even more).
- of course the individual settings need to be tweaked for each particular
system and choice of parameters and force field.
the main point is, to not get lost in trying to fix the problem where the
error occurs with all kinds of things, but to understand what is causing it
(proper visualization and studying of the files is crucial), and in the
process of understanding going back further and further until you find the
point where things go in the wrong direction, which in this case is at the
very beginning. without properly understanding what chain.py produces, you
are doomed to get it wrong.