Dear Dr. Axel Kohlmeyer and LAMMPS Users,
First, thank you Mr. Dylan Anstine for your feedback.
As previous, I am simulating uniaxial deformation of crosslinked polymer network where the nonconnected atoms are governed by LJ potential whereas connecting atoms are governed by the unbreakable FENE bonds.
I have observed that I am losing periodic bonds as the simulation box uniaxially elongates (strain rate of 0.0001 in LJ units) as shown in the below figure. The initial equilibrated crosslinked polymer network (specified in my data file which my input script reads) in its unwrapped coordinates is shown as below:

A few snapshots from the side view where the simulation box is elongating horizontally in the x-direction is shown as below where the first snapshot is the undeformed state.
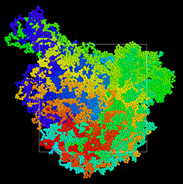


As Dylan mentioned, I do not have many bonds (in my initial equilibrated configuration) that cross the periodic boundaries in the loading direction and I am losing these bonds as the box continues to elongate until no bonds are crossing the periodic boundaries which eventually results in zero stress. Therefore, the crosslinked polymer is only following the shape of the deformed simulation box but not elastic energy is being stored in the FENE bonds.
Is this an issue regarding bad initial geometry of the equilibrated crosslinked polymer? The crosslinked polymer (subjected to NPT ensemble) is equilibrated at this final configuration.
Is there a way to keep the number of periodic bonds from the beginning and throughout the deformation process?
One way that I found out was to increase the strain rate to 0.05. At this fast strain rate, I did not lose the periodic bonds and crosslinked polymer as a whole uniaxially deformed where the FENE bonds stretched. However, if there is a way to do this with slower strain rate such as 0.0001, I would greatly appreciate for any advice.
Thank you so much for your time.
Sincerely,
Masato Koizumi
Is the total number of bonds in your system actually changing? (One way to check this is, at the end of your simulation try using write_data to create a data file. The top of that file will list the number of bonds in your system.)
This might not be the problem, but have you tried setting (increasing) the “comm_modify cutoff” distance?
https://lammps.sandia.gov/doc/comm_modify.html
Truly wacky things can happen if you neglect to specify this distance:
https://lammps.sandia.gov/threads/msg76045.html
I ran into this problem the last time I was trying to simulate a polymer which crossed periodic boundary conditions.
In the example, above, the number of bonds did not change (2 bonds), but the force from one of the bonds which was crossing the periodic boundary box was calculated using the wrong periodic image of the atoms because I forgot to specify the comm_modify cutoff.
I’ve been running into this problem when I simulate molecules containing any long bonds
(Bonds which are significantly longer than the atom diameters.)
From the documentation: “By default the ghost cutoff = neighbor cutoff = pairwise force cutoff + neighbor skin”, which is roughly within a factor of two of the sum of the atoms’ radii. In my experience, this is much more likely to happen using a coarse grained or unconventional simulation. I’m guessing this is because realistic all-atom force fields typically do not have equilibrium bond lengths which are that long.)
Anyway, I hope this helps.
It probably didn’t.
Good luck.
Andrew
If the suggestion above does not help, try starting from a simple system which does not have the problem. Then add more and more details which are specific to the simulation you want to run, until the problem re-emerges. (If you think it would help I might be able post an example of a polymer system which does not have this problem, and you could edit it.)
Dear Mr. Andrew Jewett,
First, thank you so much for your advice! In fact, the number of bonds remains constant, and, yes, I am using coarse-grained MD.
I have not tried increasing the comm_modify cutoff distance, since I am thinking that my issue here is concerned with my topology or the microscopic structure of my polymer network that I constructed (Dylan pointed this out - Thanks Dylan!)
Furthermore, my bond length is short (0.97 sigma) which is slightly shorter than the diameter of my coarse-grained bead.
Still, I deeply appreciate for your suggestions and yes when I solved my issue, I will let you know since this may potentially become a good example for the LAMMPS community to see.
Many thanks for your time!
Sincerely,
Masato Koizumi