I have attached an image below (for the visualization of my model). I want to simulate the flow of helium atoms between the Fe plates for a specific problem. There are two parallel Fe plates and a He box, as shown in the model (figure). A hybrid MEAM, LJ potential is used, MEAM for Fe-Fe, and LJ is for He-He and Fe-He. A pressure of 2.1 atm is given to the He box and a vacuum is on the other side.
#1# First I run the simulation without giving pressure (No npt condition) such as,
minimize 1.0e-2 1.0e-4 10000 10000000
velocity he create 300.0 12345 mom yes rot no
timestep 0.001
Then, the simulation runs well for all steps. But no, He atoms move in the gap of Fe sheets.
#2# When I define the pressure for He atoms such as,
minimize 1.0e-2 1.0e-4 10000 10000000
fix 1 he npt temp 300.0 300.0 0.1 iso 2.12 2.12 0.1
Then the He atoms move in the gap for the first 5800 steps, and after that, the error of the lost atom occurs.
#3# The other problem is that under the p p p boundary condition, there is also an entry of He atoms from the left side (see the image after 5800 steps. How can I stop that? (One way I put another Fe sheet on the left edge but that will not work)
I want to calculate the number of He atoms passed per second from the gap. How can I handle these errors?
Thanks for your help and suggestions.
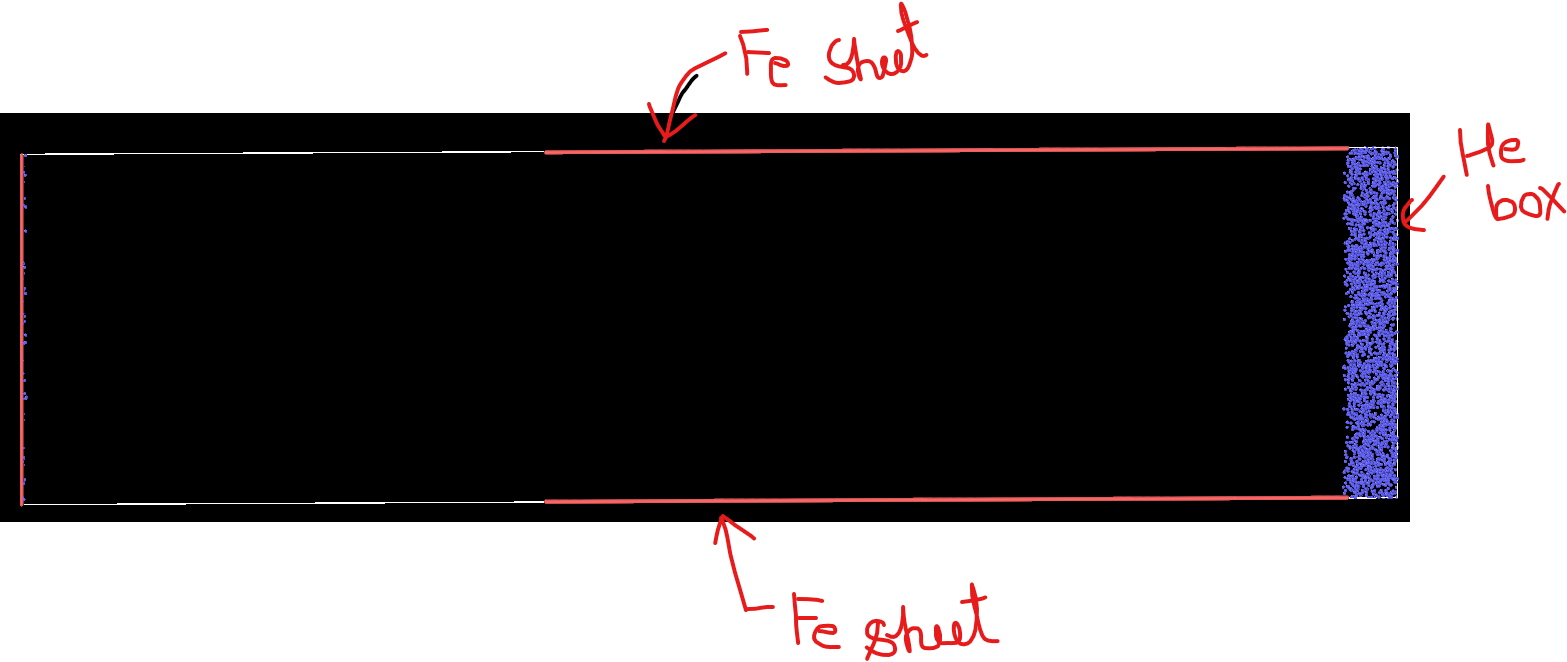

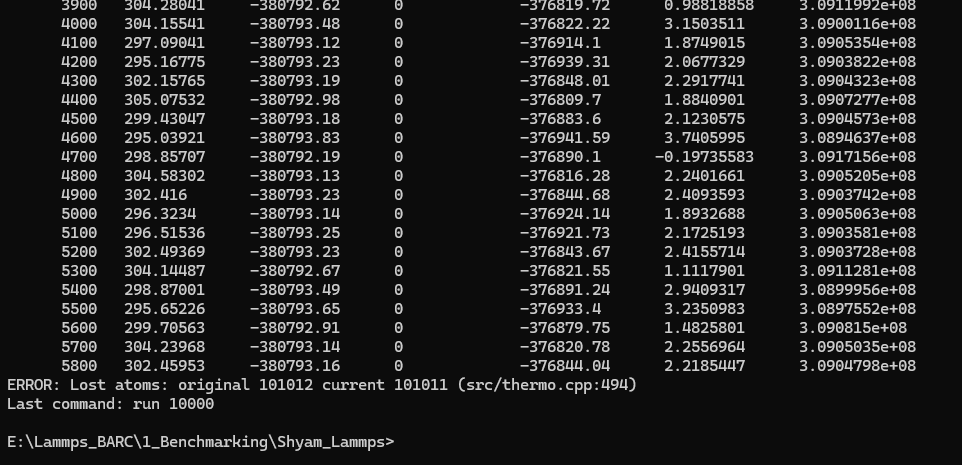
Helium_leaktightness_test.in (1.8 KB)