Dear LAMMPS users.
Hi,
I am trying to make a new compute, this compute calculates a global scalar value based on a custom per-atom vector which is created by ‘fix property/atom’.
I believe that my code works well regarding finding and accessing a custom per-atom vector (by using atom->find_custom, atom->dvector) but there is something wrong about updating values.
Below is my test run result and ‘c_elecThermalEnergy’ is my new compute. The value should be ~ 28.61 but sometimes the value is suddenly changed (completely wrong), even NaN.
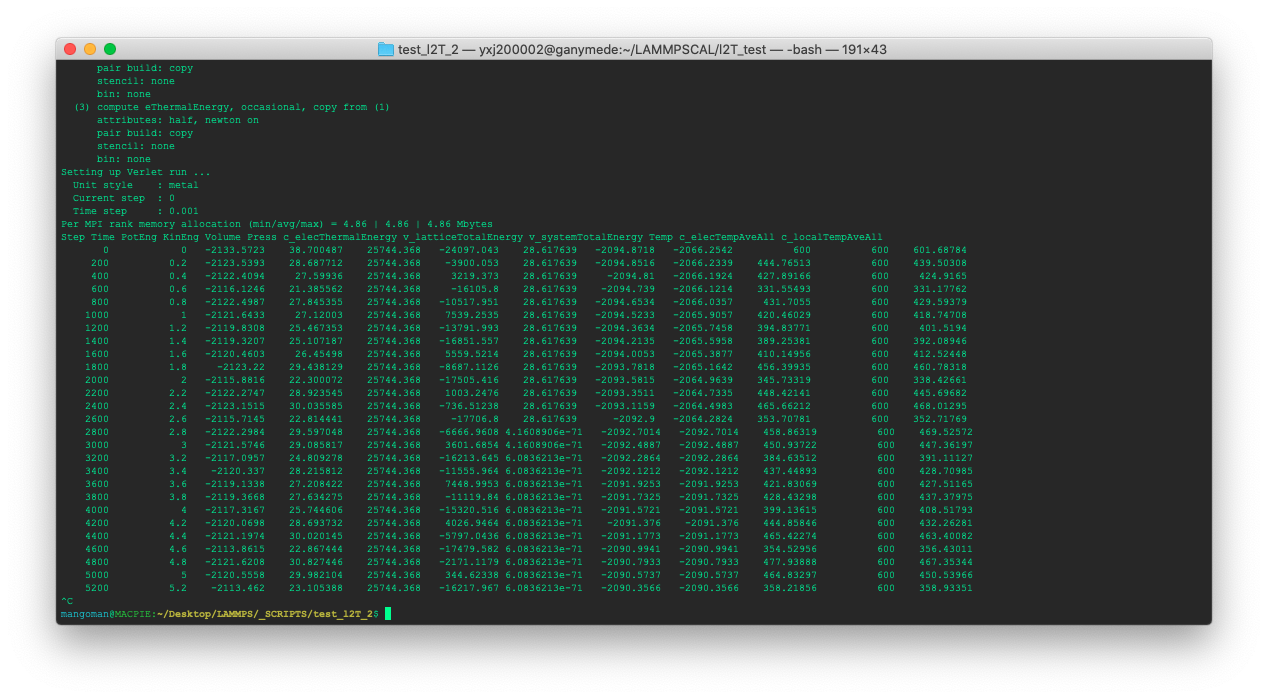
In addition to that, sometimes the value is just wrong at the start.
I am not sure what is the source of such misbehavior. Because fundamentally my compute just did a similar thing, I compared my new codes with compute_ke.cpp & compute_ke.h, but looks like that my code is just fine.
I am not fully understanding every details of LAMMPS modification, but I believe that I missed something about ‘flag’ or update routine.
Below is my new compute constructor,
ComputeElecThermalEnergy::ComputeElecThermalEnergy(LAMMPS *lmp, int narg, char **arg) :
Compute(lmp, narg, arg)
{int iarg = 0; int flag = 0; int index_custom = 0; // !! ARGUMENT !! compute ID group-ID eThermalEnergy d_ID lambda ${lambda} V ${volumePerAtom} // narg = # of arguments (counting from ID to the end) // arg = arguments (strings); start from 0 index (ID) to if (narg != 8) error->all(FLERR,"Illegal compute eThermalEnergy command"); // !!ERROR!! not enough arguments // force->numeric does interpert a string as a floating number by using 'atof'. // strcmp; compare strings. /* int iarg = 4; iarg++; */ iarg = 3; if (strstr(arg[iarg],"d_") == arg[iarg]) { // check the name of this argument started with 'd_', index_custom = atom->find_custom(&arg[iarg][2],flag); if (index_custom < 0 || flag != 1) error->all(FLERR,"Illegal compute eThermalEnergy command; the electric temperature per-atom vector does not exist."); temp = atom->dvector[index_custom]; }else{ error->all(FLERR,"Illegal compute eThermalEnergy command; the electric temperature per-atom vector should be float."); } if (strcmp(arg[4],"lambda") == 0){ lambda = force->numeric(FLERR,arg[5]); if (lambda <= 0){ error->all(FLERR,"Illegal compute eThermalEnergy command; lambda should be greater than 0"); } }else{ error->all(FLERR,"Illegal compute eThermalEnergy command"); } if (strcmp(arg[6],"V") == 0){ V = force->numeric(FLERR,arg[7]); if (V <= 0){ error->all(FLERR,"Illegal compute eThermalEnergy command; V should be greater than 0"); } }else{ error->all(FLERR,"Illegal compute eThermalEnergy command"); } // Flags scalar_flag = 1; extscalar = 1; timeflag = 1; //comm_reverse = 1;}
Below is a compute_scalar method of my new compute,
double ComputeElecThermalEnergy::compute_scalar()
{
int i;
int nlocal = atom->nlocal;
int *mask = atom->mask;
double domainSum = 0.0;/* Probably these are unnecessary parts. int *mask = atom->mask; bigint natoms = atom->natoms; */ // Don't know why, these below lines should be to make this code work. invoked_scalar = update->ntimestep; if (update->eflag_global != invoked_scalar) error->all(FLERR,"~ was not tallied on needed timestep"); /* Probably these are unnecessary parts. // grow local energy array if necessary // needs to be atom->nmax in length if (atom->nmax > nmax) { memory->destroy(energy); nmax = atom->nmax; memory->create(energy,nmax,"ee:energy"); vector_atom = energy; } */ // Calculate 'domainSum'. for (i = 0; i < nlocal; i++){ if (mask[i] & groupbit){ domainSum += (temp[i]*temp[i]); } } // Calculate 'globalSum'. MPI_Allreduce(&domainSum,&scalar,1,MPI_DOUBLE,MPI_SUM,world); scalar *= 0.5*lambda*V;return scalar;
}
If you have any comment, it would be really appreciated.
Youhwan