Thank you for your reply!
I’ve tried the one that you suggested, to copy the polyethylene and execute emc.sh build.emc
It shows the same error that there is no terminator indicating methyl.
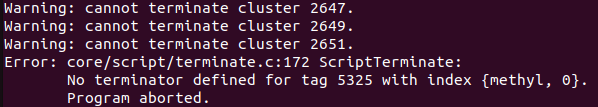
Should I change the build.emc file to fix this?
The imported polyethylene.emc is as follow:
(* EMC: Enhanced Monte Carlo simulations *)
(*
file: polyethylene.emc
author: Pieter J. in 't Veld
date: February 8, 2006
purpose: description of crystalline unit cell of polyethylene using
P.J. in 't Veld et al., Macromolecules 2006, 39, 439,
which results in atmospheric pressure at T = 402K.
*)
identity = {name → “polyethylene”};
units = {type → atomistic, length → 1, energy → 1};
variables = {alpha → 1.208435, phi → 0.733, lbond → 1.53, factor → 1.53/lbond, la → 7.7479factor, lb → 4.4626factor, lc → 2.51822*factor, sigma → 4.009044232, nsites → 4, nclusters → 2};
constants = {clusters → {polyethylene}, groups → {methylene, methyl}, sites → {ch2, ch3}};
types = {atomistic → united,
mass → {n → 2, data → {
{id → ch2, element → “C”, reference → 0, mass → 14},
{id → ch3, element → “C”, reference → 1, mass → 15}}},
standard → {
active → true,
pair → {active → true, mix → berthelot, n → 3, data → {
{ch2, ch2, 0.0934, sigma, 2.5sigma},
{ch3, ch3, 0.2265, sigma, 2.5sigma}}},
bond → {active → true, n → 2, data → {
{ch2, ch2, 899, 1.53},
{ch2, ch3, 899, 1.53}}},
angle → {active → true, n → 3, data → {
{ch2, ch2, ch2, 120, 112},
{ch2, ch2, ch3, 120, 112},
{ch3, ch2, ch3, 120, 112}}},
torsion → {active → true, n → 3, data → {
{ch2, ch2, ch2, ch2, 3, {
{0.8, 1, 0}, {0.4335, 2, 180}, {1.62, 3, 0}}},
{ch2, ch2, ch2, ch3, 3, {
{0.8, 1, 0}, {0.4335, 2, 180}, {1.62, 3, 0}}},
{ch3, ch2, ch2, ch3, 3, {
{0.8, 1, 0}, {0.4335, 2, 180}, {1.62, 3, 0}}}}}}};
groups = {
n → 2, data → {
{group → methylene, name → “METHYLENE”, terminator → false,
chemistry → “C”, nsites → 1,
sites → {
site → 0, mass → ch2, charge → 0, connects → {branch, branch},
bonds → {single, single}},
nbranches → 1,
branches → {site → 0, nconnects → 2,
connects → {{methylene, 0}, {methyl, 0}}}},
{group → methyl, name → “METHYL”, terminator → true,
chemistry → “*C”, nsites → 1,
sites → {
site → 0, mass → ch3, charge → 0, connects → {branch},
bonds → single},
nbranches → 1,
branches → {site → 0, nconnects → 1,
connects → {methylene, 0}}}}};
systems = {
n → 1, properties → {
id → 0, p → 0, v → lalblc, t → 298.15, mass → 56,
nsites → 4, nclusters → 2, geometry → {la, lb, lc, 0, 0, 0}}};
clusters = {
n → 2, data → {
{cluster → 0, id → polyethylene, head → 0, tail → 1, flags → 0b0001},
{cluster → 1, id → polyethylene, head → 2, tail → 3, flags → 0b0001}}};
sites = {
n → 4, data → {
{site → 0, cluster → 0, system → 0, mass → ch2, flags → 0b0100,
group → {methylene, 0}, connects → {1, 1, -1, -1}, p → {
la/4-lbondcos(phi)sin(alpha/2)/2,
lb/4+lbondsin(alpha/2)sin(phi)/2,
lc/4},
xtal → {{0, 0, -1}, {0, 0, 0}}},
{site → 1, cluster → 0, system → 0, mass → ch2, flags → 0b1000,
group → {methylene, 0}, connects → {0, 0, -1, -1}, p → {
la/4+lbondcos(phi)sin(alpha/2)/2,
lb/4-lbondsin(alpha/2)sin(phi)/2,
3lc/4},
xtal → {{0, 0, 0}, {0, 0, 1}}},
{site → 2, cluster → 1, system → 0, mass → ch2, flags → 0b0100,
group → {methylene, 0}, connects → {3, 3, -1, -1}, p → {
3la/4-lbondcos(phi)sin(alpha/2)/2,
3lb/4-lbondsin(alpha/2)sin(phi)/2,
lc/4},
xtal → {{0, 0, -1}, {0, 0, 0}}},
{site → 3, cluster → 1, system → 0, mass → ch2, flags → 0b1000,
group → {methylene, 0}, connects → {2, 2, -1, -1}, p → {
3la/4+lbondcos(phi)sin(alpha/2)/2,
3lb/4+lbondsin(alpha/2)sin(phi)/2,
3lc/4},
xtal → {{0, 0, 0}, {0, 0, 1}}}}};
I guess the line
connects → {{methylene, 0}, {methyl, 0}} is the problem.
