However, I do not have access to Material Studio which I believe is
needed for msi2lmp tool. Is there any other way to generate the pcff force
field for the structures?
sure, you can create those manually or with the help of some scripting
tools.
.CAR and .MDF files are are read by both msi2lmp and “EMC”. Most users use Materials Studio to create them.
In the future, moltemplate should be able to create molecules using COMPASS, CVFF, PCFF, CFF, … force fields. When that happens, hopefully you won’t need to worry about how to create .CAR and .MDF files.
Until then, as Axel said, you can make these files yourself.
You don’t need Materials Studio.
Perhaps somebody should post a tutorial how to do this.
As a first step, here is an example of .CAR and .MDF files for a molecule created by Mahdi Moshref-Javadi using materials studio (see attached picture, “751150.car” and “751150.mdf”). These files contain atomic position, charge, atom type, and bond connectivity.
Open them up in a text editor, and you’ll see they are is not very complicated.
You can make them yourself manually.
Upper-case atom names (“C1”, “C2”, …“C6”, and “O1”, “O2”, …)
are unique identifiers for each atom.
Lower-case atom names (“cp”, “c5”, “oh”, “o”,…)
are atom TYPES, which are used for looking up force-field parameters.
Note that there are multiple different types of carbon, oxygen, and hydrogen atoms, corresponding to these elements in different bonding states and in different local environments. Here is a picture of the molecule showing the atom types. To make your own file, you will have to carefully read the description of each atom type and decide which type to use at all of the atoms in your molecule, and what their partial charges should be. This is NOT trivial. (Note: You can figure out the partial charges by looking at the bond_increments section of the .FRC file and summing up all of the contributions due to all of the bonded neighbors of each atom.)
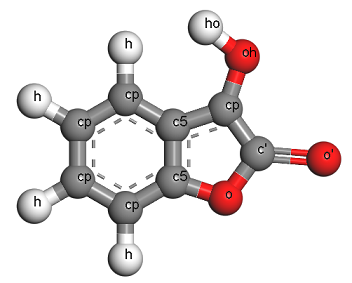
Once you have created a .CAR and an .MDF file, in principle you could use either “msi2lmp”, “EMC”, or “insight2lammps.pl” on these files.
Incidentally, running msi2lmp fails on this molecule:
msi2lmp.exe 751150 -frc ./cvff.frc
=> ERROR: Unable to find angle data for cp c5 cp
----- “insight2lammps.pl” -----
I have not tried using “EMC”, however the “insight2lammps.pl” script (distributed with “EMC” and attached to this message) generates a lammps .DATA file. Here is the syntax:
./insight2lammps.pl 751150
“751150” is the name of the file without the .car and .mdf extensions.
Make sure to copy the “cvff.frc” file into your local directory beforehand.
To change this to use the PCFF force field, edit
line 39 of “insight2lammps.pl” and replace “cvff” with “pcff”
$forcefield = “cvff”;
You will also want to copy the “pcff.frc” file into your local directory.
It is available in the tools/msi2lmp/frc_files directory in LAMMPS
(It is too big for me to include in this post.)
----- EMC -----
If somebody wants to create a tutorial for using EMC commands to convert molecules into LAMMPS format, that would be helpful.
----- bugs? -----
Does the “insight2lammps.pl” script really work with MSI force fields?
If you run the command above, it will generate a lammps .DATA file (attached “751150.data”) which nothing but zeros in the “BondBond”, “BondAngle”, and “AngleAngleTorsion” Coeffs section of the DATA file.
This behavior does not seem correct to me, but perhaps somebody more knowledgeable can clarify. Yesterday, I posted a question on the mailing list to ask for clarification on this issue:
https://sourceforge.net/p/lammps/mailman/message/35698640/
Hopefully somebody responds.
In addition to Axel’s suggestions below, I encourage you to test the molecules you create by running a simulation and comparing against known thermodynamic properties of these chemicals (eg, does it visually look like the molecule? what are its phase transition temperatures and pressures, compressibility, etc…). As the example above (with the ambiguous atom type names, partial charges, and all the zeros) suggests, there are so many ways programs like moltemplate, EMC, and msi2lmp can fail to generate the correct input files, even if when force field should work. These software tools have been tested by a limited number of people. Treat them as “beta” quality.
cheers
andrew
751150.car (1.39 KB)
751150.mdf (1.76 KB)
751150.data (18.6 KB)
