Dear Lammps Users, I am carrying out Md simulations of 2-dimensional polymers (COFs, Covalent Organic Frameworks), making use of OPLS-AA potential.
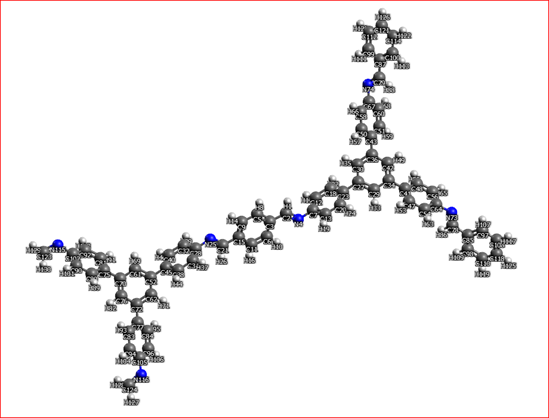
The last molecule I am studying is composed by tris-aminophenyl units (3-branched “knots”) bonded by therephtalaldehyde molecules (2-branched “linkers”). Herebelow a picture of the repeat unit.
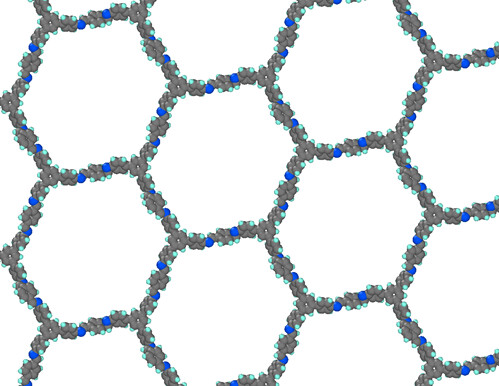
The resulting COF has hexagonal pores, see the picture below.
My work is aimed at evaluating the Young’s modulus of the COF sheet by “pulling” the atoms at the extremities of the sheet via fix move command, recording the potential energy vs strain and calculating the modulus as the second derivative of the energy-strain curve. All this after an adequate relaxation with the extremities of the sheet held firmly in place.
The issue I am facing is the following: the first runs were based on a relatively small sheet (28000 atoms, 14 x 14 “ring” framework). Everything went fine with run times up to 10 ns (1 fs step). Since the calculated modulus is usually affected by the size of the sheet, I tested a larger sheet (330000 atoms, 50 x 50 rings). Here the situation changed, in fact after about 5 ps the simulation crashed:
CPU STEP PE TEMP
2662.1358 5377 526388.81 333.52225
2662.715 5378 516104.48 347.81931
2663.2892 5379 569063.11 10578.276
2663.8633 5380 10957454 32074.255
2664.4444 5381 71826464 3.0714596e-108
ERROR on proc 0: Bond atoms 123472 129903 missing on proc 0 at step 5382 (src/ntopo_bond_all.cpp:60)
The program I am using to prepare the data file for Lammps is of course the same. I am really in trouble in finding the explanation of this behavior. Both Input and Data files are available to everybody who may be interested in having a look.
I am open to any suggestion on how I can proceed with the investigation.
Thanks a lot
Paolo
Please note that the fact that you are using the same input file does not mean that everything always go the same. Some parameters can fit for a certain system size but not another, you might be forgetting to take some implicit values into account, group definitions might be different etc. As such without the precise commands you are using it is very hard to give meaningful insights with regards to your rather complex procedure.
Good morning Germain, thanks for your kind and fast reply.
I tried to attach the Input file but it generated an error. Please accept my apolgies for being so “green” with this website. I will try to send it directly to you by mail.
Thank you so much for your interest in my troubles
Don’t send your file by email, you can copy and paste it using the Markdown syntax of the forum by putting three backticks (```) before and after your text. That will keep the file readable and not format the text.
That way anyone with better knowledge can provide you some help.
units real
boundary p p p
atom_style full
pair_style lj/cut/coul/cut 10 12
bond_style harmonic
angle_style harmonic
dihedral_style opls
improper_style cvff
atom_modify map array
pair_modify table 0
read_data d-COFPA-BRU51x51-00
minimize 0.001 0.00001 100 1000
group UP id 321302 321303 321304 321305 321306 321307 321309 321311 321312
group DOWN id 2 3 4 5 6 7 9 11 12 13 15 18 20 21 23 25 27
fix moveUP UP move linear 0 0 0
fix moveDOWN DOWN move linear 0 0 0
group NUC subtract all UP DOWN
fix mynvt NUC nvt temp 300 300 100
velocity NUC create 300 887723
neighbor 2 bin
neigh_modify delay 0 every 1 check yes page 100000 one 10000
thermo 1000
thermo_style custom cpu step pe temp
dump mydump all custom 1000 dump-COFPA-BRU51x51-10 id type x y z
# restart 1000 rest1 rest2
run 1000000
write_restart restart-COFPA-BRU51x51-10
write_data dat-COFPA-BRU51x51-10
Dear Germain, please note that the the group id UP and DOWN lines do no list all the atoms which should be involved. They are very many and I copied only a few of them.
These atoms are located at the extremities of the sheet in the y axis direction. They are not involved in the issue.
The two atoms which loose their mutual bonds belong to two adjacent repeat units, so I guess the issue might be there.
This input is rather complicated to understand what you are doing. I can only make a couple of rather vague comments:
First concerning format, backticks ` are not the same character as ticks '. But that’s not really important here.
The fix move linear 0 0 0 commands are useless as is. You equivalently keep your atoms in groups UP and DOWN outside of the nvt integrated group NUC. But you are using period boundary conditions in every direction. This is odd to me. I do not understand how you can pull on your COF sheet that way. A more common way of doing with PBC is using fix deform.
The definition of UP and DOWN is also cumbersome. I think you could have better result with a smart combination of groups using either type or region commands. As is, it is nearly impossible to understand.
I would suggest using velocity zero all linear after your velocity create command and a temperature compute using NUC group to ensure that the temperature that the fix monitor is the proper temperature of the integrated atoms.
Most importantly as far as I can see on your figures (since I have no data file to confirm), you appear to have hydrogen atoms in your model. If I remember correctly, hydrogen bonding has to be constrained in OPLS-AA forcefield. Please have a look at the fix shake command to learn how to do so.
As a side note, the reason for these constraints is the very different vibration frequencies of hydrogen covalent bonds and other covalent bonds. An alternative would be to severely decrease the integration timestep but that means order of magnitude longer simulations.
These comments are mostly based on guesses since I don’t know how your system is generated, the parameters you used or the actual geometry of the system but they already point to problems you might have to look into. I cannot pinpoint your exact problem and why it did not happen with smaller systems but that is something you should figure out through more detailed investigation.
Please note that first your kinetic energy very suddenly increases, then also the potential energy. That is an indication of either a bad force field parameter set (which is not sufficiently repulsive to keep charges sufficiently apart to avoid a “charge catastrophe”) or - in this case more likely - too large a timestep. A 1fs timestep is only reliable if you use fix shake to constrain all bonds involvin hydrogen atoms.
With a larger system you increase the probability that atoms get too close through thermal fluctuations (and thus trigger the bad behavior) in two ways: 1) with more atoms, there are more possible locations that can misbehave and 2) with a larger system you can have larger fluctuations.
The best method to share (large) files and work around the “novice forum member” restriction (which is enabled to discourage spammers) is to upload the files to Dropbox, Google Drive, One Drive or similar and then create and provide a shareable link.
This should be p p f for a 2-d polymer system you describe.
For parallel efficiency when running with MPI, it is also advisable to add: processors * * 1
You will have to also check whether it is more accurate to use lj/cut/coul/long with kspace_style pppm and kspace_modify slab
I strongly agree with @Germain that you should be using change_box or fix deform instead of “pulling at the edges”. That is much more bulk-like and thus avoids unphysical boundary effects, which can be worse than the finite size effects you try to avoid with running a larger system. The challenge is, of course, that you now need to create bonds across the periodic boundary (and ignore LAMMPS’ warning about inconsistent image flags which cannot be avoided for this kind of system).
Why increase neigh_modify one? You have a system with fewer neighbors than a 3d bulk molecular system. The default settings in LAMMPS are fully sufficient for that with standard density and cutoff <=15 Angstrom. This change in settings just wastes memory. If anything, you can probably make “one” smaller (you can see the typical number of neighbors per atom in the run summary at the end of a run.
The temperature here is tainted because it includes atoms that have no kinetic energy. Also the atoms bound to the immobile atoms will have a damped kinetic energy and thus contribute to the non-bulk behavior of the boundary area. So even the temperature of the fix nvt group is not 100% accurate and the thermostat leading to a somewhat tainted total temperature.
Axel, Germain, thank you so much for your comments and recommendations. I will make the best use I can of them to improve my original script.
I am pretty conscious that my “Lammps style” is very rough: my fields of knowledge, if any, are located elsewhere, so the fact of having competent people available to give assistance is a real boon.
Paolo
Dear Axel, Germain, further on my problem of simulation crash I would like to ask again for your comments.
I introduced the fix shake command and some other modifications as per your recommendations. This fix involves all Hydrogen atom types, as well as all bond and angle types in which a H atom is involved.
The simulation crashes again, typically because of lost atoms of an angle. By looking with OVITO the molecule behavior frame by frame I noticed that the beginning of the crash starts with an H atom flying away from its C companion.
The fix shake command instructions say:
“In LAMMPS, only small clusters of atoms can be constrained. This is so the constraint calculation for a cluster can be performed by a single processor, to enable good parallel performance. A cluster is defined as a central atom connected to others in the cluster by constrained bonds. LAMMPS allows for the following kinds of clusters to be constrained: one central atom bonded to 1 or 2 or 3 atoms, or one central atom bonded to 2 others and the angle between the 3 atoms also constrained. This means water molecules or CH2 or CH3 groups may be constrained, but not all the C-C backbone bonds of a long polymer chain.”
In my case all H atoms of the 2D framework (330000 atoms in total) are involved in the fix, so I am wondering if my case infringes the “small clusters” rule, which might explain the persistence of the problem.
Any comment would be as usual of the greatest help. Thanks for your patience.
The SHAKE fix should be applied to carbon hydrogen bonds. As such CH bonds should be constant and this should not happen.
In your case it makes no sense to not use the all group. Shake takes into account bond and angle types so you should include both hydrogen and carbon atoms.
Dear Germain, sorry to come back to your last comment so late.
I do not really understand what exactly you do mean with: " In your case it makes no sense to not use the all group. Shake takes into account bond and angle types so you should include both hydrogen and carbon atoms."
Have I to specify both H and C types (after the “t” constraint, I assume) involved in the bonds and angles I woul like to “freeze”? Would it not be sufficient to specify bond and angle types with the “b” and “a” constraints?
I suggest you take a careful reading of the shake manual because it seems you overlooked at least two things.
The type constraint constraints all atoms that are bonded to an atom of the specified type. In OPLS only hydrogen bonds are constrained. So you do not have to specify C types. I would also recommend to use the b constraint with the required carbon-hydrogen bond types which is, in my opinion, more clear and specific in the case of OPLS.
You’re said that all H atoms [...] are involved in the fix. It was unclear to me if they were the only atoms involved or not. It makes no sense to restrict the application to the fix to hydrogen atoms because the documentation states clearly that: For all constraints, a particular bond is only constrained if both atoms in the bond are in the group specified with the SHAKE fix.
Applying shake to a group containing only hydrogen atoms would then be useless in your case. You should likely use the group all.
Hi Germain, thanks a lot for your quick reply, not to mention your patience. I went through the documentation more carefully. hope to have properly understood the matter. I will modify the input as per your last recommendations. Let’s see if my simulation may run properly this time.
Dear Germain, I am very sorry to bother you again with my difficulties. Hope your patience is not totally exhausted.
Following the last posts, I tried to adopt all the suggestions I received. I prepared a new input script (see below) wiith the aim of testing its functioning. This time there is no “pulling”: I will leave hopefully to a later phase the utilization of the fix deform command. This script is just “pinning” in their position the atoms of the upmost and downmost rows of the 2D molecule. The run is set at 1 ns.
Here is what happens:
when the temperature is set at 1 °K (fix mynvt NUC nvt temp 1.0 1.0 100; velocity all create 1.0 369157), everything works fine and the simulation stops at 1 ns without any issue. Potential energy comes to a fairly constant value,as well as temperature.
when the temperature is set at 300 °K (the desired temperature for my study), after few ps the screen starts showing: “WARNING: Shake determinant < 0.0 (src/RIGID/fix_shake.cpp:1721)”. This situation lasts unchanged up to half an hour or so, when I stop the simulation. This behavior occurred during all the several runs I tried.
One additionaal piece of information: in fix shake I specified after the b the C-H bond types and after the a the H-C-C angle types.
That’s that: I am really lost, so, as usual, any comment or explanation would be of the greatest value
Paolo
COFPA51x51 FILENAME in-COFPA51x51-200 10/07/2023
EQUILIBRATION
RUN 1000 ps
processors * * 1
units real
boundary p p f
atom_style full
pair_style lj/cut/coul/cut 10 12
bond_style harmonic
angle_style harmonic
dihedral_style opls
improper_style cvff
atom_modify map array
pair_modify table 0
read_data d-COFPA51x51-30
minimize 0.001 0.00001 100 1000
group UP id 321302 321303 321304 321305 321306 321307 .......
group DOWN id 2 3 4 5 6 7 9 11 12 ......
group NUC subtract all UP DOWN
fix mynvt NUC nvt temp 300.0 300.0 100
velocity all create 300.0 1258
velocity all zero linear
fix myshake all shake 0.0001 20 0 b 1 5 8 11 a 1 2 5 9 12 14
neighbor 2 bin
neigh_modify delay 0 every 1 check yes page 100000
thermo 1000
thermo_style custom cpu step pe temp
dump mydump all custom 1000 dump-COFPA51x51-200 id type x y z
restart 1000 rest1 rest2
run 1000000
write_restart restart-COFPA51x51-200
write_data dat-COFPA51x51-200
This is an indication that there may be something wrong with your system topology (i.e. geometry plus bond/angle assignments). This message would happen when you try to constrain a geometry where you have angles that are close to 180 degrees at which point the constraint equations can no longer be solved due to numerical instability from the limitations of floating point math.
Please note that your post is starting a new topic. It is considered a bad habit to follow up a discussion of one topic with a different one in the same thread. It can be very misleading to people reading the discussions later or search the archives for help. In the future please post new questions that are unrelated to an existing topic as a new topic. Thanks.
Axel, my apologies. I simply put this post as a reply to an existing one by ermain. THis post is related to the issues I am facing with a simulation of the very same molecule. So, in my view, this post was related to the same topic. In the future I will try to avoid similar mistakes.
Axel, the system topology is generated always in the same way.
More specifically, the data file used for the simulation at 1 °K as described in my previous post, which run smoothly for 1 ns, is exactly the same data file used for the simulation at 300 °K, which gave rise to the “WARNING: Shake determinant < 0.0 (src/RIGID/fix_shake.cpp:1721)" issue.
Also the input scripts of both simulations are exactly the same, the changes are limited to the numerical values of the temperature (1 vs. 300).
In synthesis I have difficulties to understand where are the errors in a system topology which is always the very same, in one case (run at 1 °K) giving no problems and in the second making troubles (run at 300 °K).
I am puzzled by the fact that by changing the temperature of the simulation, the behavior of the simulation is radically different.
Following what I hope to have understood of fix shake, I specified the H-C bonds types and the H-C-C angles “to be reset to their equilibrium lengths and angular values”. My objective is apply the shake algorithm to C-H atoms as per Germain’s recommendation and, maybe incorrectly I assumed that both C-H bonds and H-C-C angles must be specified.