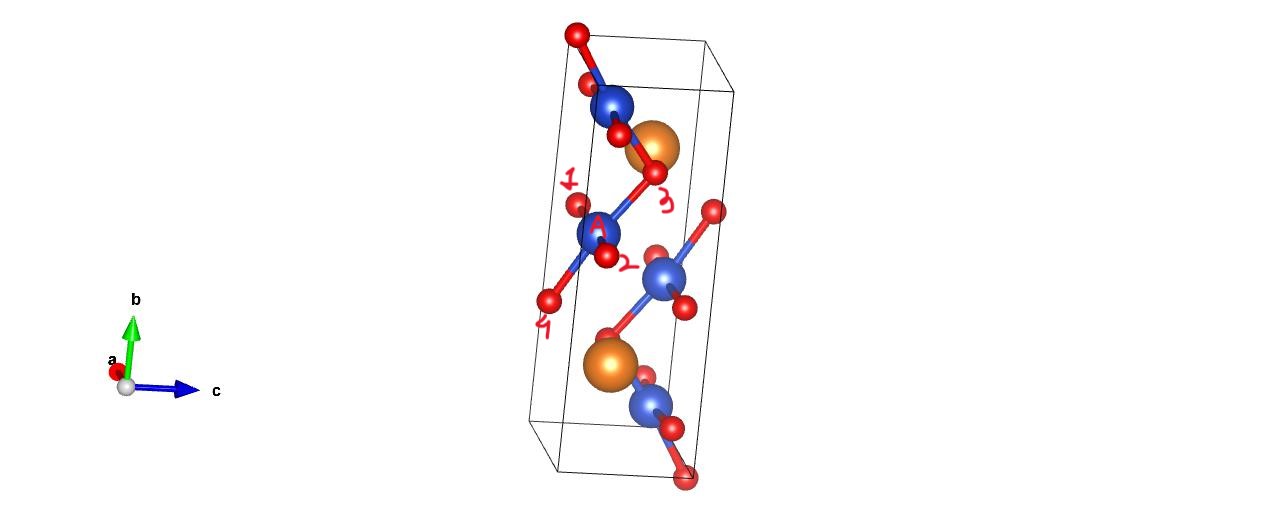
I wish to calculate the distance of all the atoms from a particular atom(atom A) in my cell. But I also want to include the atoms that might lie just beyond the periodic unit cell boundary.( In figure, besides distance of A with 1,2,3 i also want distance with 4). I am using get_distance from ASE, but it will not include atom outside(atom 4) the boundary. I am printing the nearest atoms to A to confirm this. What can be done to consider this?
The code I am using is(Atom A is “Cu” and atom 1,2,3,4 is “O”) :
import numpy as np
from ase.io import read,write
from ase.geometry import get_distances
from ase.neighborlist import NeighborList,natural_cutoffs,build_neighbor_list,first_neighbors,get_connectivity_matrix,neighbor_list
structure= read("1985check.cif")
atom_count=structure.get_chemical_symbols()
#atom_count=supercell.get_chemical_symbols()
for i in range(len(atom_count)):
if(atom_count[i]=='Cu'):
index=i
break
neighbor_list=[]
j_list=[]
for j in range(len(atom_count)):
if(j!=index):
neighbor_list.append(structure.get_distance(index,j,mic=True))
j_list.append(j)
keys= sorted(range(len(neighbor_list)), key=lambda k: neighbor_list[k])
check_list=[]
for c in range(len(keys)):
#print(structure.get_chemical_symbols()[j_list[keys[c]]])
check_list.append(structure.get_chemical_symbols()[j_list[keys[c]]])
The output of check_list which writes the atom distances in increasing order is [‘O’,‘O’,‘O’, ‘Cu’…] while I expect [‘O’,‘O’,‘O’, ‘O’…]
The cif file is Dropbox - 1985check.cif - Simplify your life