Dear Lammps users,
I am currently running MD simulation on nanoporous graphene, particularly mechanical tension on the membrane, in order to extract the stress-strain diagram.
For that I am using displace_atoms command that was used in Simon Gravelle Tutorial on CNT and Graphene sheet.
Surprisingly the membrane doesn’t t deform as it should,since to edges where I apply the strain get dissociated during the running and the rest of the membrane stays fixed. as show this picture :
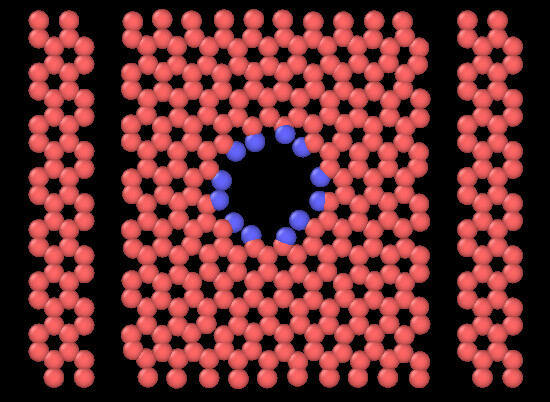
I am wondering if it’s due to the potential (I am using CH airebo) or due to the command itself.
Does anyone encounter the same issue or have an idea how to solve this issue?
below is my input.
data1.data (98.1 KB)
input1.in (2.1 KB)
Thank you