Hello all,
I am trying to simulate the metal-water interface based on the Helmholtz model using ReaxFF.
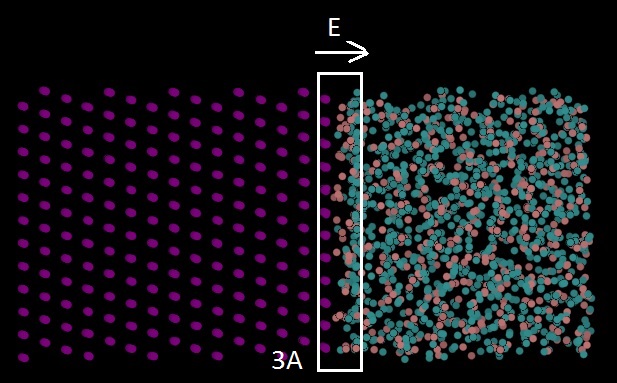
I applied the electric field 3 A0 (as the electrical double layer is 3A0 to 5A0 in Helmholtz model) from the surface of the metal which affected the surface metal atoms (nickel) and a couple of water layers. The electric field is constant as the potential in Helmholtz model is linear. I modified the electrostatic energy to take into account electronegativity difference and charge conservation.
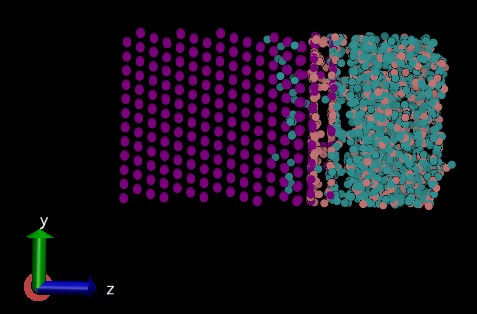
I put 4 A0 on the nickel side as vacuum and set up a group that does not include the bottom 3 layers of nickel atoms and did time integration only on those. Attachment 1 is my input file.
The problem is that when I included additional electrostatic forces Fi=qi*Eexternal using “efield” command on that 3 A0, my system is getting separated (most of the water molecules go to the right side, attachment 2). But I don’t have this problem (attachment 3) when I apply the electric field using “efield” command on all the system, or when I don’t apply this force (deactivate “efield” command).
I am not sure if I should modify my model or am using the efield command inappropriately. I appreciate if you let me your ideas.
Regards,
Hossein,
in.nirun (1.92 KB)

Hello all,
I am trying to simulate the metal-water interface based on the Helmholtz
model using ReaxFF.
your description below is too confusing to me to make sense of it, but
i would like to point out one important fact:
the fix efield command doesn't really implement an electric field; it
implements a charge dependent per atom force. that is equivalent to an
interaction with an electric field in vacuum at infinite dilution of
atoms and for non-polarizable atoms.
with a dense system, and particularly with a system using charge
equilibration, fix efield is an extremely crude approximation. the
atoms are not polarized as they should and the charge equilibration
doesn't know anything about the external field (but should).
axel.
Axel,
Thank you for your reply.
I’m sorry it was confusing. Here I try to make it more clear:
I am going to apply electric field as shown in attached figure to see the metal-water interactions. Electric field Eel contributes an additional energy term E(q,Eel) associated with the charge on the ions as below (J. Phys. Chem. A 2012,116,11796-11805):
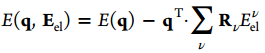
I applied this equation in the LAMMPS code, and also changed the electronegativity of the ions by taking into account Eel :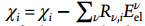 .
.
Finally, I included additional electrostatic forces 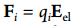 using “efield” command for that 3 A0 in the first attached figure.
using “efield” command for that 3 A0 in the first attached figure.
As I mentioned earlier, when I do it the system is getting separated after awhile (most of the water molecules go to the right side, attachment 2). But I don’t have this problem when I apply the force on the whole system using “efield” command (attachment 3).
Still I couldn’t figure out why my system is getting separated. Maybe I need to apply the force on the whole system.
Thank you,
Hossein,
But I don’t have this problem (attachment 3) when I apply the electric field
Axel,
Thank you for your reply.
I'm sorry it was confusing. Here I try to make it more clear:
I am going to apply electric field as shown in attached figure to see the
metal-water interactions. Electric field Eel contributes an additional
energy term E(*q*,*Eel*) associated with the charge on the ions as below
(J. Phys. Chem. A 2012,116,11796-11805):
!image.png|265x53
I applied this equation in the LAMMPS code, and also changed the
electronegativity of the ions by taking into account Eel :[image: Inline
image 4] .
Finally, I included additional electrostatic forces [image: Inline image
3]using "efield" command for that 3 A0 in the first attached figure.
As I mentioned earlier, when I do it the system is getting separated after
awhile (most of the water molecules go to the right side, attachment 2).
But I don't have this problem when I apply the force on the whole system
using "efield" command (attachment 3).
the paper you are referencing shows a test of the necessary
modifications. have you been able to reproduce this test accurately?
also, have you been able to reproduce the results w/o the application of
the electric field?
what you are doing is a modification of an extremely complex force field.
people on the mailing list here do not have time to debug your code. that
is something you have to do yourself and it should be done with extreme
care considering the complexity of what you are trying to do.
Still I couldn't figure out why my system is getting separated. Maybe I
need to apply the force on the whole system.
this is not scientific approach to solve a problem (make arbitrary changes
until it looks right). you have to _understand_ why you have to make
certain choices in a particular way. besides, if anybody, the logical
people to contact are the authors of the paper that you are trying to
apply/reproduce.
axel.
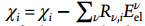
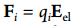
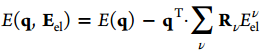{kind=link}