Hi all,
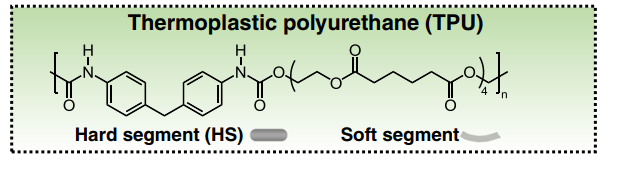
I’m a very new user to EMC, and I’m trying to build a TPU polymer using opls-aa force field and generate lammps input files. Below is the chemical formula of my polymer:
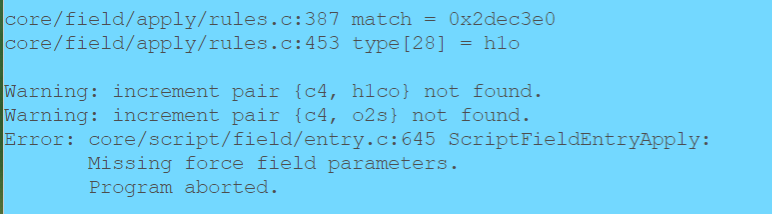
But when I tried to use EMC, I got the following errors:
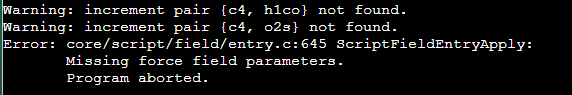
I found it really confusing, and I have checked the 2024/opls-aa.prm, and what I found were:
c4: sp3 carbon
h1co: hydrogen in aldehyde and formamide
o2s: oxygen in esters
I found it really confusing, is that opls-aa does not have the corresponding parameters? while these atoms and the chemical environments seem to be quite common to me? I don’t know if it is wrong with my esh file:
ITEM OPTIONS
project TPU
field opls-aa
auto true
ntotal 130
density 0.1
replace true
ITEM END
# Groups
ITEM GROUPS
mono *COC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)NC1=CC=C(CC2=CC=C(NC*=O)C=C2)C=C1,1,mono:2
methyl *C,1,mono:1,1,mono:2
ITEM END
# Clusters
ITEM CLUSTERS
poly alternate 1
ITEM END
# Polymers
ITEM POLYMERS
poly
1 mono,2,methyl,2
ITEM END
The SMILES I write is COC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)CCCCC(=O)OCCOC(=O)NC1=CC=C(CC2=CC=C(NC=O)C=C2)C=C1, no idea if this is the problem, really confused. Could anyone help me please? Many thanks!