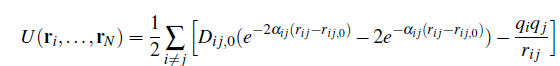b) The fact is that it is not exactly a sum, the contribution added to the morse potential counteracts coulomb interactions, in fact it has opposite sign and that . I could create a new pair_coul/long changing the sign and use that. I would use coul/cut since the point of the coulomb correction is to restore the bond length between the atoms so it is a short range one.
you can change the sign of how the charges are applied with a trick: using fix adapt with a fixed scale factor of -1.0 should subtract the coulomb interaction instead of adding it.
here is a modified version of the melt input, that adds coul/cut to lj/cut
units lj
atom_style charge
lattice fcc 0.8442
region box block 0 10 0 10 0 10
create_box 2 box
create_atoms 1 box
mass * 1.0
set group all type/fraction 2 0.5 526223499
set type 1 charge 1.0
set type 2 charge -1.0
velocity all create 3.0 87287
pair_style hybrid/overlay lj/cut 4.0 coul/cut 4.0
pair_coeff * * lj/cut 1.0 1.0
pair_coeff * * coul/cut
neighbor 0.5 bin
neigh_modify every 2 delay 0 check yes
thermo_style custom step temp pe evdwl ecoul etotal
thermo 50
fix 1 all nve
fix 2 all adapt 1 pair coul/cut scale * * v_qscale
variable qscale equal 0
run 0 post no
variable qscale equal 1
run 0 post no
variable qscale equal -1
run 0 post no
run 250 post no
you can see, how the contribution from coulomb changes as the scale variable changes.
[…]
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 8.737 | 8.737 | 8.737 Mbytes
Step Temp PotEng E_vdwl E_coul TotEng
0 3 -7.1092599 -7.1092599 0 -2.6103849
Loop time of 9.53674e-07 on 1 procs for 0 steps with 4000 atoms
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 8.737 | 8.737 | 8.737 Mbytes
Step Temp PotEng E_vdwl E_coul TotEng
0 3 -6.9824594 -7.1092599 0.12680048 -2.4835844
Loop time of 7.15256e-07 on 1 procs for 0 steps with 4000 atoms
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 8.737 | 8.737 | 8.737 Mbytes
Step Temp PotEng E_vdwl E_coul TotEng
0 3 -7.2360604 -7.1092599 -0.12680048 -2.7371854
Loop time of 9.53674e-07 on 1 procs for 0 steps with 4000 atoms
Setting up Verlet run …
Unit style : lj
Current step : 0
Time step : 0.005
Per MPI rank memory allocation (min/avg/max) = 8.737 | 8.737 | 8.737 Mbytes
Step Temp PotEng E_vdwl E_coul TotEng
0 3 -7.2360604 -7.1092599 -0.12680048 -2.7371854
50 1.7116093 -5.3280547 -5.0561652 -0.27188947 -2.7612825
100 1.7334776 -5.4030907 -4.9967851 -0.40630552 -2.8035243
150 1.8002483 -5.6083344 -4.9535691 -0.65476534 -2.9086371
200 1.8695844 -5.8020733 -4.8911685 -0.91090481 -2.9983978
250 1.9599233 -6.0949476 -4.837561 -1.2573866 -3.1557976
Loop time of 3.43038 on 1 procs for 250 steps with 4000 atoms
Total wall time: 0:00:03
axel.